Buzzwords
- Klinefelter (47,XXY): Tall male, gynecomastia, small/firm testes, infertility, ↑FSH/LH.
- Turner (45,XO): Short stature, webbed neck, shield chest, streak ovaries, bicuspid aortic valve, coarctation.
- Androgen Insensitivity (46,XY): Phenotypic female, blind-ending vagina, absent uterus, scant pubic hair, cryptorchid testes.
- 5α-Reductase Deficiency (46,XY): Ambiguous genitalia at birth, virilization at puberty (“penis at 12”), ↑Testosterone:DHT ratio.
- Müllerian Agenesis (46,XX): Primary amenorrhea w/ normal secondary sex characteristics, absent uterus, normal ovaries.
- 21-Hydroxylase Deficiency (CAH):
- 46,XX: Ambiguous genitalia (virilization).
- Both: Salt-wasting (hypotension, ↓Na+, ↑K+), precocious puberty.
- Buzzword Lab: ↑ 17-hydroxyprogesterone.
- Swyer Syndrome (46,XY Gonadal Dysgenesis, loss-of-function SRY): Phenotypic female, has a uterus, “streak gonads” (must be removed), no puberty (no breasts/menses).
- Kallmann Syndrome: Anosmia (can’t smell) + hypogonadism (delayed puberty); ↓GnRH, ↓FSH, ↓LH.

| Feature | Müllerian agenesis | Androgen insensitivity syndrome | 5-alpha reductase deficiency |
|---|---|---|---|
| Karyotype | 46,XX | 46,XY | 46,XY |
| Pathogenesis | Absent or hypoplastic müllerian ducts | Androgen resistance due to X-linked androgen receptor mutation | Deficient conversion of testosterone to dihydrotestosterone (DHT) |
| Hormone levels | Normal estrogen & testosterone | ↑ Testosterone & estrogen (aromatization) | ↑ Testosterone, ↓ DHT |
| Internal Sex Organs | No Uterus/Upper Vagina; Ovaries present | No Uterus/Upper Vagina; Testes present | Testes present; Male ducts (Wolffian) |
| External genitalia | Normal female | Normal female | Often ambiguous at birth, virilizes at puberty |
| Breasts | Normal | Normal | Minimal to absent (Testosterone is enough to suppress breast) |
| Axillary & pubic hair | Normal | Minimal to absent | Sparse to normal (varies) |
| Wolffian structures | Absent | Absent | Partial development |
| Gender identity | Female | Usually female | May change to male at puberty |
| Timing of diagnosis | Usually at adolescence (primary amenorrhea) | May be diagnosed at birth, puberty, or adulthood | Often at birth (ambiguous genitalia) or puberty |
| Risk of malignancy | No increased risk | ↑ Risk of gonadal tumors after puberty | ↑ Risk of gonadal tumors |
Klinefelter syndrome
Epidemiology
- One of the most common causes of male hypogonadism
Etiology
- Associated with advanced maternal age
Pathophysiology
- Karyotype: 47,XXY is most common (can be 48,XXXY, etc.).
- Mechanism: Caused by meiotic nondisjunction of sex chromosomes.
- One Barr body (inactivated X chromosome) is present.
- Primary Hypogonadism: Testicular dysgenesis leads to failure of both seminiferous tubules and Leydig cells. t
- Dysgenesis of Seminiferous Tubules: ↓ Inhibin B loss of feedback inhibition ↑ FSH.
- Abnormal Leydig Cell Function: ↓ Testosterone ↑ LH ↑ Estrogen.
Clinical features
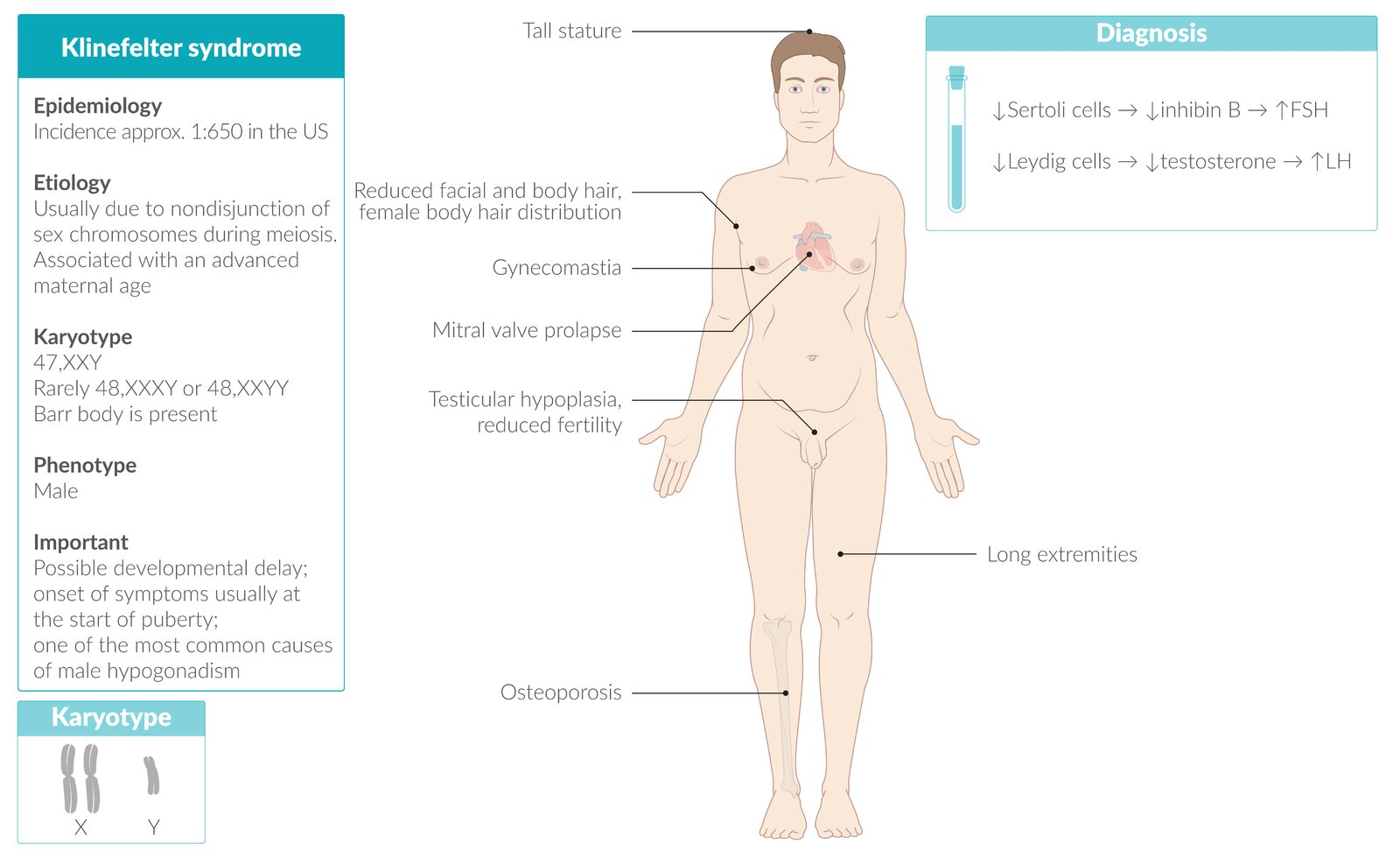
- Eunuchoid growth pattern: tall, slim stature with long extremities (Growth plate closure is delayed )
- Gynecomastia
- Testicular Atrophy: Small, firm testes (due to fibrosis).
- Kallmann syndrome has small, soft testes. t
- Possible developmental delay
- Neurocognitive dysfunction (impaired executive function and memory, decreased intelligence)
- Associated disorders
- Mitral valve prolapse
- Increased risk of breast and testicular cancer
- Due to increased estrogen
Turner syndrome
Pathophysiology
- Chromosomal nondisjunction → chromosome X monosomy/mosaicism → impaired ovarian development → malfunctioning streak gonads with connective tissue instead of normal germ cells → estrogen and progesterone deficiencies
- The absence of a second X chromosome disrupts X-linked survival signals, causing primordial germ cells to undergo accelerated apoptosis, leading to near-total germ cell loss by birth. Without germ cells, ovarian structures (e.g., follicles) fail to develop, and regressing gonadal ridges are replaced by fibrous connective tissue, forming non-functional streak gonads.
- Karyotype
- Meiotic nondisjunction (most often in paternal gametes) → complete sex chromosomal monosomy (45,XO; no Barr body)
- Barr body: The inactive X chromosome present in all female somatic cells. Appears as a small, dark-staining spot at the periphery of the nucleus. Consists of tightly-packed, transcriptionally-inactive, heterochromatin.
- Mitotic nondisjunction of an embryonic cell → sex chromosomal mosaicism (45,XO/46,XX) → mild phenotypic expression
- Meiotic nondisjunction (most often in paternal gametes) → complete sex chromosomal monosomy (45,XO; no Barr body)
Clinical features
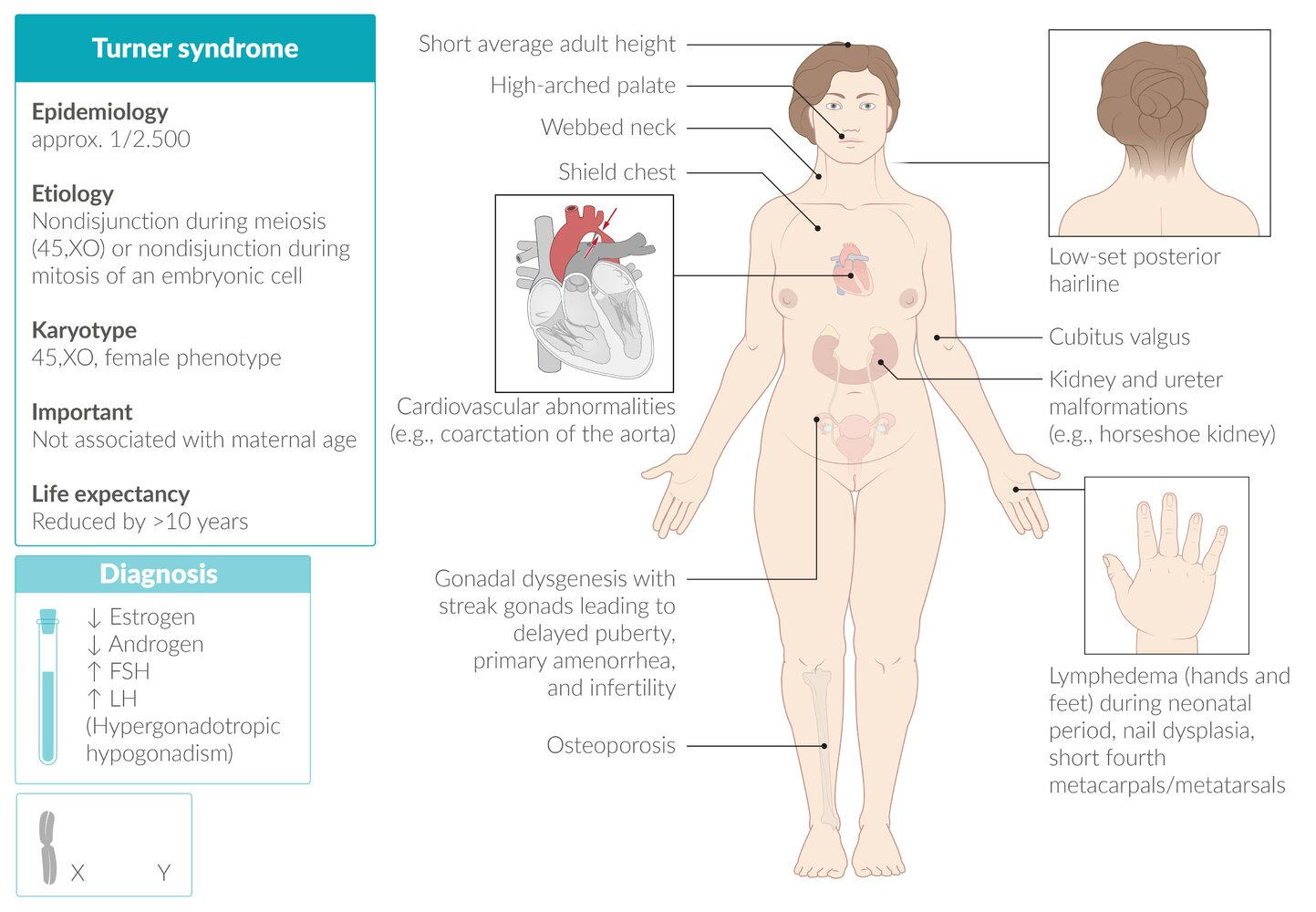
- Short stature (most consistent finding).
- Primary amenorrhea (most common cause in patients with a uterus).
- Physical Exam:
- Webbed neck (pterygium colli) and low posterior hairline.
- Shield chest with widely spaced nipples.
- Lymphedema of hands and feet, especially in newborns.
- Shortened 4th metacarpals.
- Intelligence is typically normal, though some patients may have challenges with spatial reasoning.
- Associated Conditions (Very High-Yield)
- Cardiovascular (50% of patients):
- Bicuspid aortic valve (most common cardiac defect).
- Coarctation of the aorta (preductal).
- ↑ Risk of aortic dissection and aneurysm. c
- Renal:
- Horseshoe kidney is the most common anomaly. t
- Other structural malformations can occur.
- Endocrine:
- Ovarian dysgenesis (“streak ovaries”) leads to premature ovarian failure.
- ↑ Risk of hypothyroidism (Hashimoto’s thyroiditis).
- ↑ Risk of Type 2 Diabetes Mellitus.
- Other:
- Recurrent otitis media and sensorineural hearing loss.
- Increased risk for autoimmune diseases (e.g., celiac disease).
- Cardiovascular (50% of patients):
Tip
Most patients with Turner syndrome have normal intelligence.
Kallmann syndrome
Etiology
Hypogonadotropic hypogonadism with hyposmia/anosmia
Pathophysiology
- Core defect: Failure of GnRH-releasing neurons to migrate from the olfactory placode to the hypothalamus during embryonic development.
- ↓ GnRH → ↓ pituitary secretion of FSH and LH → ↓ testosterone in male individuals and ↓ estrogen in female individuals
- The defective migration also involves the olfactory axons, leading to aplasia or hypoplasia of the olfactory bulbs, which causes the impaired sense of smell.
Clinical features
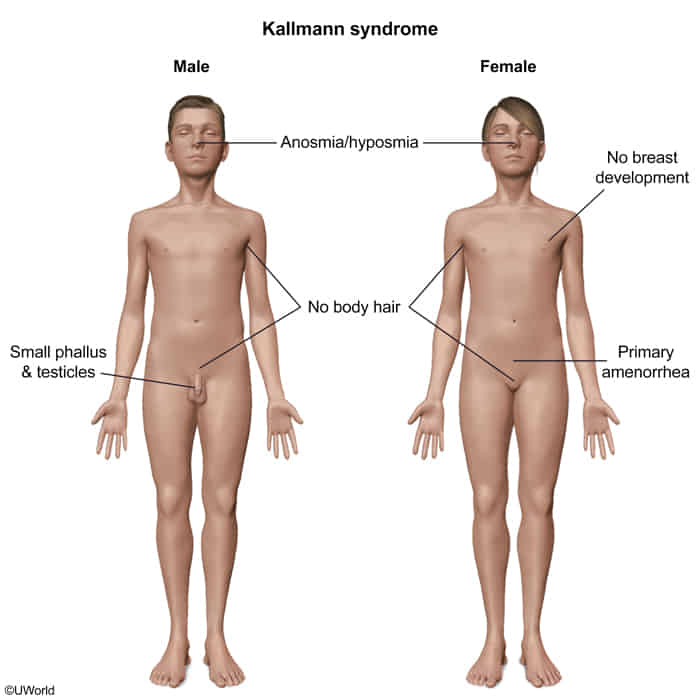
- Hallmark Finding: The combination of delayed or absent puberty with an impaired or absent sense of smell is pathognomonic.
- Reproductive Features (due to GnRH deficiency):
- Both sexes: Absent or incomplete puberty, infertility.
- Males: May present in infancy with micropenis or cryptorchidism. Post-puberty, they exhibit small testes (<4 mL), low libido, and reduced muscle mass.
- Females: Present with primary amenorrhea and lack of breast development.
- Non-Reproductive Features:
- Midline defects: Cleft lip or palate.
- Renal anomalies: Unilateral renal agenesis is a classic association.
- Neurologic: Bimanual synkinesis (mirror movements of the hands), cerebellar ataxia, hearing impairment.
- Skeletal: Dental agenesis.
Androgen insensitivity syndrome
- Pathophysiology
- Dysfunction of the AR in the hypothalamus and pituitary leads to loss of feedback inhibition of gonadotropin-releasing hormone (GnRH), FSH, and LH. This results in the following hormonal findings:
- GnRH induces increased LH secretion, which leads to increased testosterone production in the testes.
- FSH secretion is increased by GnRH but suppressed by inhibin from the seminiferous tubules and is often normal.
- Estrogen, which is derived by aromatization of testosterone, may be normal or elevated.
- Dysfunction of the AR in the hypothalamus and pituitary leads to loss of feedback inhibition of gonadotropin-releasing hormone (GnRH), FSH, and LH. This results in the following hormonal findings:
- Diagnostics
- Clinical presentation
- Before puberty: ↑ testosterone
- After puberty: ↑ LH, ↑ estrogen, and normal/↑ testosterone levels (no virilization)
- Genetic testing
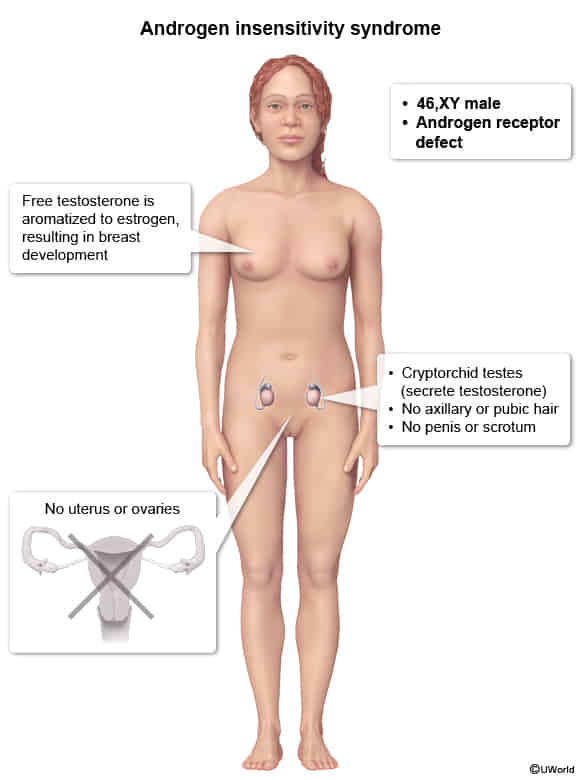 Tall stature: due to delayed fusion of the epiphyseal growth plates.
Tall stature: due to delayed fusion of the epiphyseal growth plates.
SRY Gene Translocation
-
SRY Gene Function: The Sex-determining Region Y (SRY) gene, located on the Y chromosome, is the primary trigger for male sex determination. It codes for a protein (testis-determining factor) that initiates the development of testes from the bipotential embryonic gonads.
-
Patho/Etiology: Abnormal crossing-over during paternal meiosis moves the SRY gene from the Y to the X chromosome.
- 46,XX Testicular DSD: Individual is XX but has the SRY gene, leading to a male phenotype.
- 46,XY Gonadal Dysgenesis (Swyer Syndrome): Individual is XY but has a deleted/mutated SRY gene, leading to a female phenotype due to lack of testes development.
-
Presentation:
- 46,XX Male: Presents with small testes, gynecomastia, and infertility. Phenotypically male.
- 46,XY Female (Swyer): Presents with primary amenorrhea and lack of secondary sex characteristics. Has a uterus but streak gonads. Phenotypically female.
-
Dx:
- Karyotype shows discordance between genotype (e.g., 46,XX) and phenotype (male).
- FISH or PCR confirms SRY gene presence on X-chromosome (in 46,XX male) or its absence/mutation (in 46,XY female).
- Hormones: Elevated LH/FSH. Low testosterone (XX male) or low estrogen (XY female).
-
Mgmt:
- 46,XX Male: Testosterone replacement for virilization.
- 46,XY Female (Swyer): Estrogen/progestin HRT for female development. Prophylactic gonadectomy is crucial due to high risk of gonadoblastoma.
-
Key Complications:
- Infertility (universal).
- Gonadoblastoma risk in the streak gonads of 46,XY Swyer Syndrome.
- Psychosocial distress.
Treatment
- Hormone Therapy: Often essential.
- For 46,XY DSDs (like Androgen Insensitivity Syndrome, 5-alpha reductase deficiency, Swyer Syndrome): Estrogen or testosterone may be used depending on the specific condition and gender identity. Gonadectomy (removal of testes) is common in AIS and Swyer Syndrome due to cancer risk.
- For 46,XX DSDs (like Congenital Adrenal Hyperplasia, Aromatase Deficiency, 46,XX Testicular DSD): Corticosteroids are vital for CAH. Estrogen is used for conditions like Aromatase Deficiency and XX Gonadal Dysgenesis. Testosterone may be used for 46,XX Testicular DSD.
- For Sex Chromosome DSDs: Growth hormone and estrogen for Turner Syndrome (45,XO); testosterone for Klinefelter Syndrome (47,XXY).
- Specific Interventions:
- Kallmann Syndrome: Hormone replacement to induce puberty.
- Congenital Adrenal Hyperplasia (CAH): Lifelong corticosteroids to manage hormone imbalances.
- Ovotesticular DSD: Individualized hormone therapy and surgery based on gonadal tissue and gender identity.
- Double Y Syndrome (47,XYY): No direct cure; treatment is supportive (e.g., speech therapy).