Generalized Proximal Tubule Defect
- Fanconi Syndrome
- Pathophysiology: A generalized reabsorption defect in the Proximal Convoluted Tubule (PCT). Impaired transport of glucose, amino acids, HCO₃⁻, phosphate, and other solutes. Leads to significant urinary wasting of these substances.
- Key Features:
- Normal serum glucose despite glucosuria is a major clue. (Plasma glucose is tightly regulated by mechanisms (eg, insulin, glucagon) independent of renal reabsorption.)
- Metabolic Acidosis (Type 2 RTA): Due to urinary loss of bicarbonate (HCO₃⁻).
- Hypophosphatemia: Leads to rickets in children and osteomalacia in adults.
- Aminoaciduria, Polyuria.
- Causes:
- Hereditary: Wilson disease, cystinosis, galactosemia.
- Acquired: Multiple myeloma (light chains), lead poisoning, and nephrotoxic drugs (e.g., expired tetracyclines, ifosfamide, certain antiretrovirals).
Renal tubular acidosis (RTA)
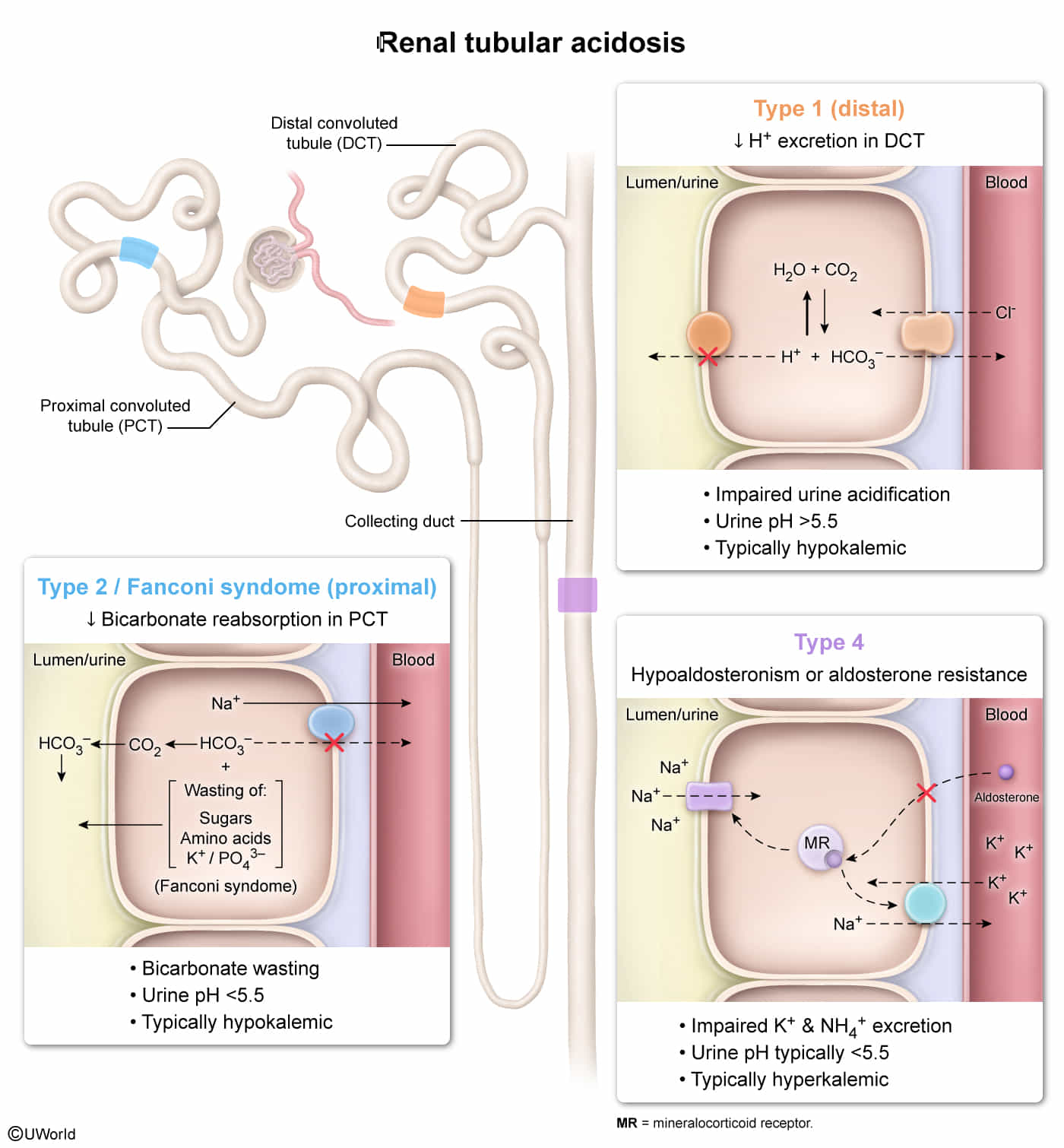
- In RTA, there is a normal anion gap metabolic acidosis in patients with normal or almost normal renal function.
- Basically all present with low pH and hypokalemia (except type 4)
- Think H+ and K+ antagonize each other during excretion (due to Na+/K+ vs Na+/H+), increased excretion of one leads to decreased excretion of the other.
- Renal tubular acidosis is caused by defects in the tubular transport of HCO3- and/or H+.
- Most forms of RTA are asymptomatic; rarely, life-threatening electrolyte imbalances may occur.
Remember these alphabetically

Renal Tubular Acidosis (RTA) is a group of disorders causing a non-anion gap, hyperchloremic metabolic acidosis with a relatively preserved glomerular filtration rate (GFR). The core issue is a failure of the renal tubules to properly handle acid excretion or bicarbonate (HCO3⁻) reabsorption.
Type 1 (Distal) RTA
- Pathophysiology: Defect in H+ secretion by α-intercalated cells in the distal tubule/collecting duct. This leads to an inability to acidify the urine.
- Etiology:
- Autoimmune diseases: Sjögren syndrome, Rheumatoid Arthritis (RA), SLE.
- Medications: Amphotericin B, lithium.
- Hereditary causes.
- Clinical Presentation:
- Hypokalemia.
- Recurrent calcium-phosphate kidney stones and nephrocalcinosis due to alkaline urine (pH > 5.5), hypercalciuria, and hypocitraturia.
- In children: failure to thrive, rickets.
- Diagnosis:
- Hallmark: Urine pH > 5.5 despite systemic metabolic acidosis.
- Serum: Normal anion gap acidosis, hypokalemia.
- Management: Alkali replacement with sodium bicarbonate or potassium citrate to correct acidosis and prevent stone formation.
Type 2 (Proximal) RTA
- Pathophysiology: Impaired HCO3⁻ reabsorption in the proximal convoluted tubule (PCT).
- Etiology:
- Fanconi Syndrome: A generalized PCT defect causing phosphaturia, glucosuria, and aminoaciduria.
- Plasma glucose is tightly regulated by mechanisms (eg, insulin, glucagon) independent of renal reabsorption. Therefore, serum glucose remains normal.
- Medications: Carbonic anhydrase inhibitors (e.g., acetazolamide), expired tetracyclines.
- Multiple myeloma (light chain toxicity to tubules).
- Fanconi Syndrome: A generalized PCT defect causing phosphaturia, glucosuria, and aminoaciduria.
- Clinical Presentation:
- Hypokalemia.
- Bone disease (osteomalacia/rickets) due to phosphate wasting and chronic acidosis.
- Symptoms related to Fanconi syndrome if present.
- Diagnosis:
- Serum: Normal anion gap acidosis, hypokalemia.
- Urine pH: Initially high (>5.5) due to bicarbonaturia. Once serum HCO3⁻ is significantly depleted, less bicarbonate is filtered and the distal tubule can still acidify the urine, leading to a urine pH < 5.5.
- Fractional excretion of HCO3⁻ >15% during a bicarbonate infusion test.
- Management: Requires large doses of alkali (e.g., sodium bicarbonate) and potassium supplementation. Thiazide diuretics may be used.
Type 4 (Hyperkalemic) RTA
- Pathophysiology: Aldosterone deficiency or resistance, leading to impaired Na+ reabsorption and K+/H+ secretion in the collecting duct. The resulting hyperkalemia inhibits ammonia (NH3) synthesis, further reducing acid excretion.
- Etiology:
- Diabetic nephropathy (hyporeninemic hypoaldosteronism) is a classic cause.
- Medications: ACE inhibitors, ARBs, NSAIDs, K+-sparing diuretics (e.g., spironolactone), TMP-SMX.
- Adrenal insufficiency (Addison’s disease).
- Clinical Presentation:
- Usually mild, asymptomatic acidosis.
- Hyperkalemia is the hallmark finding and can cause cardiac arrhythmias.
- Diagnosis:
- Serum: Normal anion gap acidosis, hyperkalemia.
- Urine pH < 5.5 (distal acidification ability is intact).
- Management:
- Treat the underlying cause and stop offending drugs.
- Low-potassium diet.
- Loop or thiazide diuretics can be used to manage hyperkalemia.
- Fludrocortisone (a mineralocorticoid) may be used in cases of aldosterone deficiency if the patient is not volume-overloaded.
| Feature | Type 1 (Distal) | Type 2 (Proximal) | Type 4 (Hyperkalemic) |
|---|---|---|---|
| Defect | ↓ H+ secretion | ↓ HCO3⁻ reabsorption | Aldosterone deficiency/resistance |
| Serum K+ | Low | Low | High |
| Urine pH | > 5.5 | Variable (< 5.5 once acidotic) | < 5.5 |
| Key Association | Kidney stones, autoimmune dz | Fanconi syndrome, multiple myeloma | Diabetes, ACE inhibitors |
Tip
Type 1 (Distal) RTA
- High Urine pH (>5.5): The distal H+ pump is broken, so acid (H+) cannot be secreted. The urine is always alkaline.
- Low K+ (Hypokalemia): The body wastes K+ in the urine to try and conserve H+. Also, the H+/K+ pump that reabsorbs K+ is impaired.
Type 2 (Proximal) RTA
- Variable Urine pH (<5.5 when severely acidotic): The proximal tubule leaks bicarbonate (HCO3⁻), initially making urine alkaline. Once serum HCO3⁻ is very low, there’s none left to leak, and the normal distal tubule can successfully acidify the urine.
- Low K+ (Hypokalemia): The lost HCO3⁻ in the tubule acts as a diuretic, washing K+ out into the urine.
Type 4 (Hyperkalemic) RTA
- Low Urine pH (<5.5): The problem is aldosterone failure, not broken H+ pumps. However, the resulting hyperkalemia prevents the kidney from making ammonia (NH3), which is needed to buffer the acid. Urine is acidic, but total acid excretion is low.
- High K+ (Hyperkalemia): This is the root cause. Without aldosterone, the kidney cannot secrete K+, so it builds up in the blood.
Mixed RTA (type 3)
- Type 1 RTA with HCO3- wasting
Disorders Mimicking Diuretic Use
- Bartter Syndrome
- Pathophysiology: Defect in the Na⁺-K⁺-2Cl⁻ cotransporter (NKCC) in the thick ascending limb of the loop of Henle. Mimics chronic loop diuretic use.
- Key Features:
- Presents in childhood.
- Hypokalemia, Metabolic Alkalosis.
- Hypercalciuria (high urine Ca2+) because the disrupted ion gra dient impairs paracellular Ca2+ reabsorption.
- Normal to low blood pressure.
- Gitelman Syndrome
- Pathophysiology: Defect in the Na⁺-Cl⁻ cotransporter (NCC) in the Distal Convoluted Tubule (DCT). Mimics chronic thiazide diuretic use.
- Key Features:
- Milder, often presents in adolescence or adulthood.
- Hypokalemia, Metabolic Alkalosis.
- Hypocalciuria (low urine Ca2+) and Hypomagnesemia. The latter is a key differentiator from most other tubulopathies.
- Normal to low blood pressure.
Disorders Mimicking Hyperaldosteronism
Both present with hypertension, hypokalemia, and metabolic alkalosis, but have low aldosterone levels.
- Liddle Syndrome
- Pathophysiology: Gain-of-function mutation in the epithelial Na⁺ channel (ENaC) in the collecting tubule. Leads to excessive Na⁺ reabsorption and K⁺ secretion, independent of aldosterone.
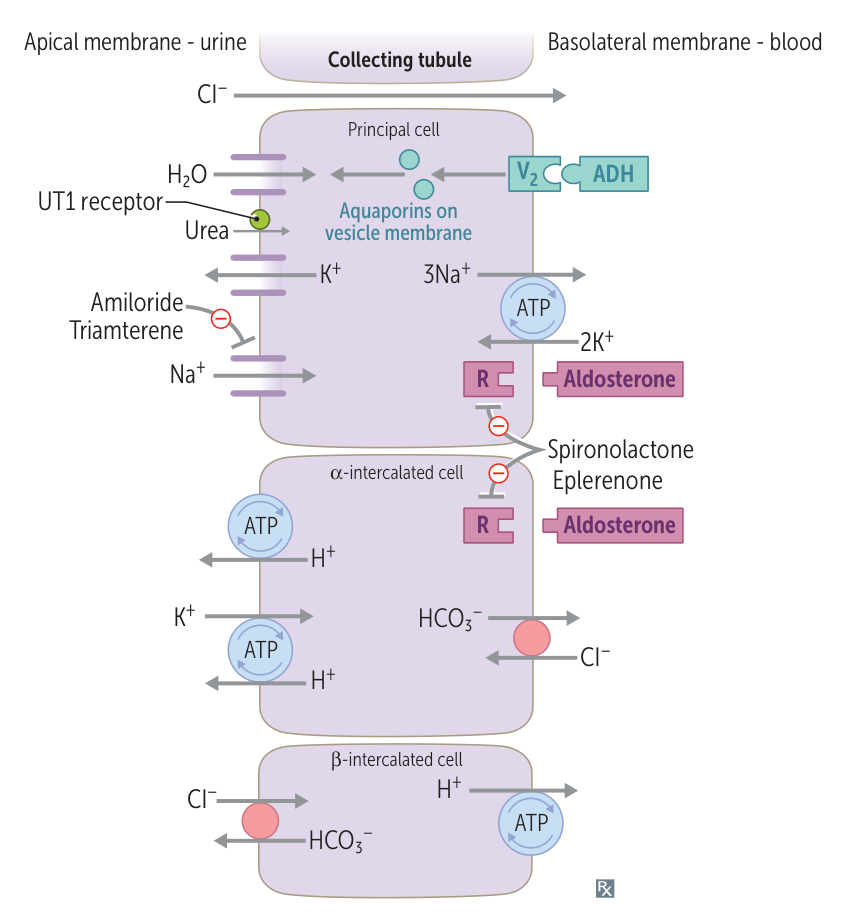
- Key Features:
- Autosomal dominant inheritance.
- Presents as early-onset hypertension.
- ↓ Aldosterone and ↓ Renin due to feedback from hypertension and volume expansion.
- Treatment: Amiloride or Triamterene (ENaC blockers).
- Pathophysiology: Gain-of-function mutation in the epithelial Na⁺ channel (ENaC) in the collecting tubule. Leads to excessive Na⁺ reabsorption and K⁺ secretion, independent of aldosterone.
- Syndrome of Apparent Mineralocorticoid Excess (SAME)
- Pathophysiology: Deficiency of 11β-hydroxysteroid dehydrogenase type 2. This enzyme normally inactivates cortisol to cortisone in mineralocorticoid receptor-expressing cells. Without it, excess cortisol activates the mineralocorticoid receptor.
- Key Features:
- Presents like Liddle syndrome (hypertension, hypokalemia, metabolic alkalosis).
- ↓ Aldosterone and ↓ Renin.
- Can be acquired from consuming licorice (glycyrrhetinic acid), which inhibits the enzyme.
- Treatment: Corticosteroids (e.g., dexamethasone) to suppress endogenous cortisol production; potassium-sparing diuretics.