- Amyloid: insoluble protein or protein fragments
- Amyloidosis: extracellular aggregation and subsequent deposition of amyloid in various organs
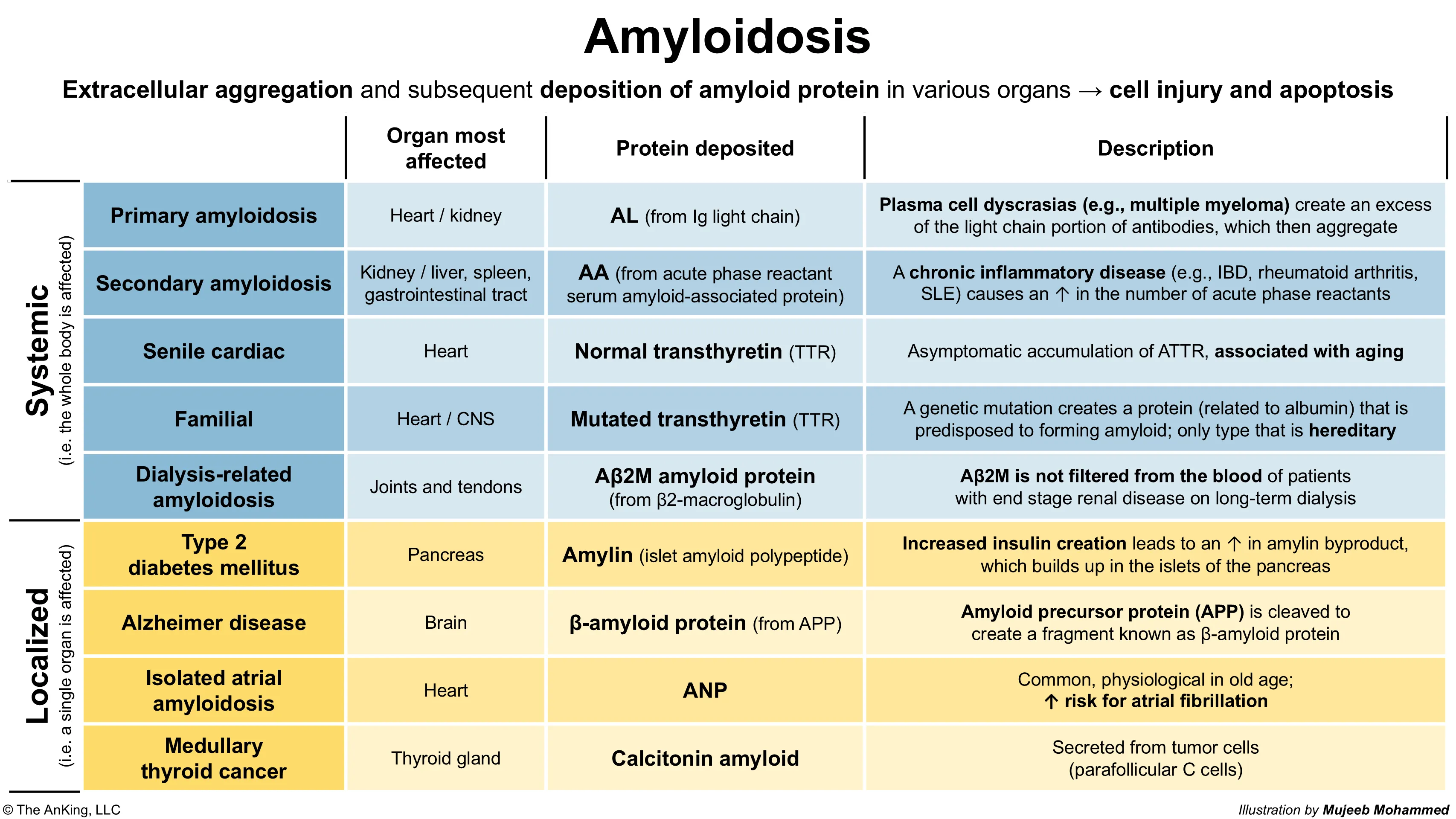

AL amyloidosis (primary amyloidosis)
Most common form of amyloidosis
- Pathophysiology/Etiology: Clonal plasma cell disorder (e.g., multiple myeloma, MGUS) produces abnormal immunoglobulin light chains (L in AL) that misfold and deposit in tissues. This is the most common type of systemic amyloidosis. t
- Clinical Presentation: Highly variable. Often presents with nephrotic syndrome (proteinuria), restrictive cardiomyopathy (HFpEF, arrhythmia), peripheral/autonomic neuropathy, hepatosplenomegaly, and macroglossia. “Raccoon eyes” (periorbital purpura) is a classic but less common sign.
- Diagnosis:
- Screening: Serum/urine protein electrophoresis (SPEP/UPEP) with immunofixation and serum free light chain (FLC) assay to detect a monoclonal protein.
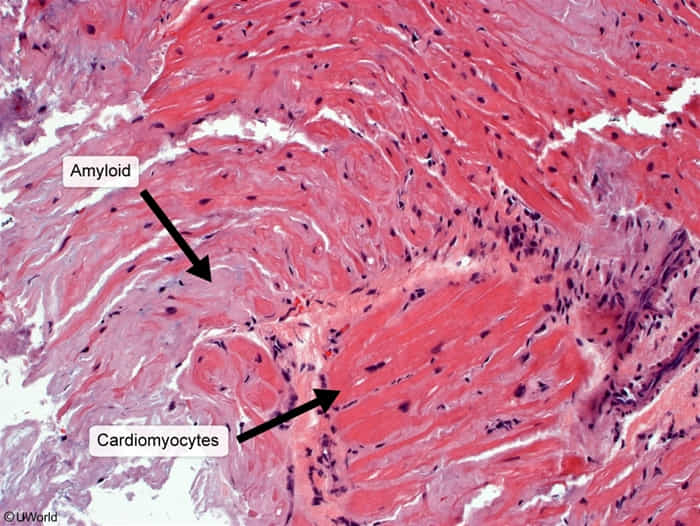
- Gold Standard: Tissue biopsy (abdominal fat pad, bone marrow, or affected organ) showing apple-green birefringence on Congo red staining under polarized light.
- Typing: Mass spectrometry or immunohistochemistry on the biopsy is crucial to confirm the amyloid is from light chains.
- Management/Treatment: Aims to eliminate the underlying plasma cell clone.
- First-line: Chemotherapy, often a combination like Dara-CyBorD (daratumumab, cyclophosphamide, bortezomib, dexamethasone).
- Autologous stem cell transplant (ASCT) may be an option for eligible patients.
AA amyloidosis (secondary amyloidosis)
- Pathophysiology/Etiology: Occurs secondary to chronic inflammatory or infectious conditions (e.g., rheumatoid arthritis, IBD, chronic infections like osteomyelitis or tuberculosis, FMF). Chronic inflammation leads to persistently high levels of serum amyloid A (SAA), an acute-phase reactant, which is then cleaved and deposited as amyloid A (AA) protein.
- Clinical Presentation: Primarily affects the kidneys, causing proteinuria and nephrotic syndrome, which can progress to ESRD. Hepatosplenomegaly is also common.
- Diagnosis: Same biopsy findings as AL (Congo red positive). Diagnosis requires excluding AL and identifying an underlying chronic inflammatory state.
- Management/Treatment: Treat the underlying inflammatory or infectious disease to reduce SAA production. For example, colchicine for Familial Mediterranean Fever (FMF).
Tip
Amyloidosis should always be considered in patients who present with kidney, liver, or GI involvement in the setting of chronic inflammatory and/or infectious disease.
ATTR (Transthyretin) Amyloidosis
- Pathophysiology/Etiology: Deposition of transthyretin (TTR), a protein mainly produced by the liver.
- Hereditary (ATTRv): Caused by a mutation in the TTR gene, leading to an unstable protein.
- Wild-Type (ATTRwt): Occurs in older individuals (>65) due to age-related instability of normal TTR protein (formerly called senile systemic amyloidosis).
- Clinical Presentation:
- Cardiac: Restrictive cardiomyopathy is the hallmark, especially in ATTRwt.
- Neurologic: Peripheral and autonomic neuropathy are common, especially in ATTRv.
- Other: Bilateral carpal tunnel syndrome often precedes cardiac symptoms by years.
- Cardiac: Restrictive cardiomyopathy is the hallmark, especially in ATTRwt.
- Diagnosis:
- In a patient with suspected cardiac amyloidosis, if monoclonal protein tests are negative, a technetium-99m-pyrophosphate (PYP) scan is highly specific for ATTR cardiac amyloidosis (shows strong myocardial uptake).
- Genetic testing is required to differentiate between ATTRv and ATTRwt.
- Management/Treatment:
- TTR Stabilizers: (e.g., Tafamidis, Diflunisal) prevent TTR from misfolding.
- TTR Silencers: (e.g., Patisiran, Inotersen) are RNA-interference agents that reduce the production of TTR by the liver.
- Supportive care for heart failure is crucial.
Dialysis-Related Amyloidosis (Aβ2M)
- Pathophysiology/Etiology:
- Deposition of β2-microglobulin (β2M), a normal component of the MHC Class I molecule. t
- Normally freely filtered at the glomerulus, then reabsorbed and broken down by the proximal convoluted tubule (PCT).
- In ESRD, β2M is not adequately filtered by the kidneys or by older, low-flux hemodialysis membranes.
- Chronically elevated serum levels of β2M lead to its deposition as amyloid fibrils, primarily in osteoarticular structures.
- Deposition of β2-microglobulin (β2M), a normal component of the MHC Class I molecule. t
- Clinical Presentation:
- Almost exclusively seen in patients on long-term dialysis (typically >5-10 years).
- Presentation is primarily musculoskeletal.
- Key Manifestations:
- Carpal tunnel syndrome (often the first sign). t
- Scapulohumeral periarthritis (destructive arthritis of the shoulder, causing chronic shoulder pain and limited motion).
- Destructive spondyloarthropathy (affecting the spine).
- Bone cysts and pathologic fractures.
- Diagnosis:
- Primarily a clinical diagnosis based on characteristic symptoms in a long-term dialysis patient.
- Biopsy of affected tissue (e.g., synovial membrane) shows classic Congo red staining with apple-green birefringence.
- Immunohistochemistry or mass spectrometry is required to confirm the protein is β2M.
- Management/Treatment:
- Prevention: Use of modern, high-flux dialysis membranes or hemodiafiltration, which are more effective at clearing β2M.
- Definitive Treatment: Successful kidney transplantation is curative, as it restores normal renal clearance of β2M.
- Symptomatic management for arthropathy (e.g., NSAIDs with caution, physical therapy, joint surgery).