| Disorder | Key Gene / Locus | Primary Skin Finding | Primary CNS Finding | ”Can’t Miss” Association |
|---|---|---|---|---|
| Sturge-Weber | Sporadic (GNAQ) | Port-Wine Stain (V1/V2) | Leptomeningeal Angioma (“Tram-track” calcifications) | Glaucoma |
| Tuberous Sclerosis | TSC1/TSC2 | Ash-leaf spots, Angiofibromas | Cortical Tubers, Subependymal giant cell astrocytomas (SEGAs) | Cardiac Rhabdomyoma, Renal Angiomyolipoma |
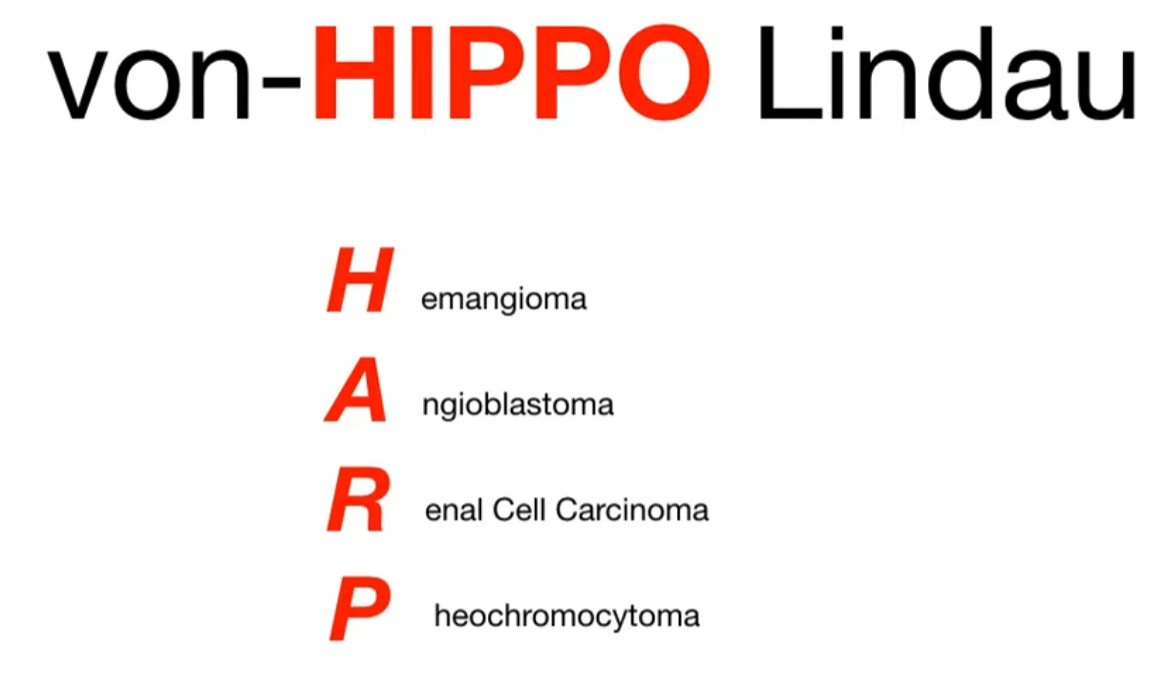
| von Hippel-Lindau | VHL (Chr 3) | (Minimal skin findings) | Hemangioblastomas (retina, cerebellum) | Renal Cell Carcinoma, Pheochromocytoma |
| NF1 | NF1 (Chr 17) | Café-au-lait spots, Cutaneous neurofibromas Axillary freckling | Optic Glioma | Lisch nodules (iris hamartomas) |
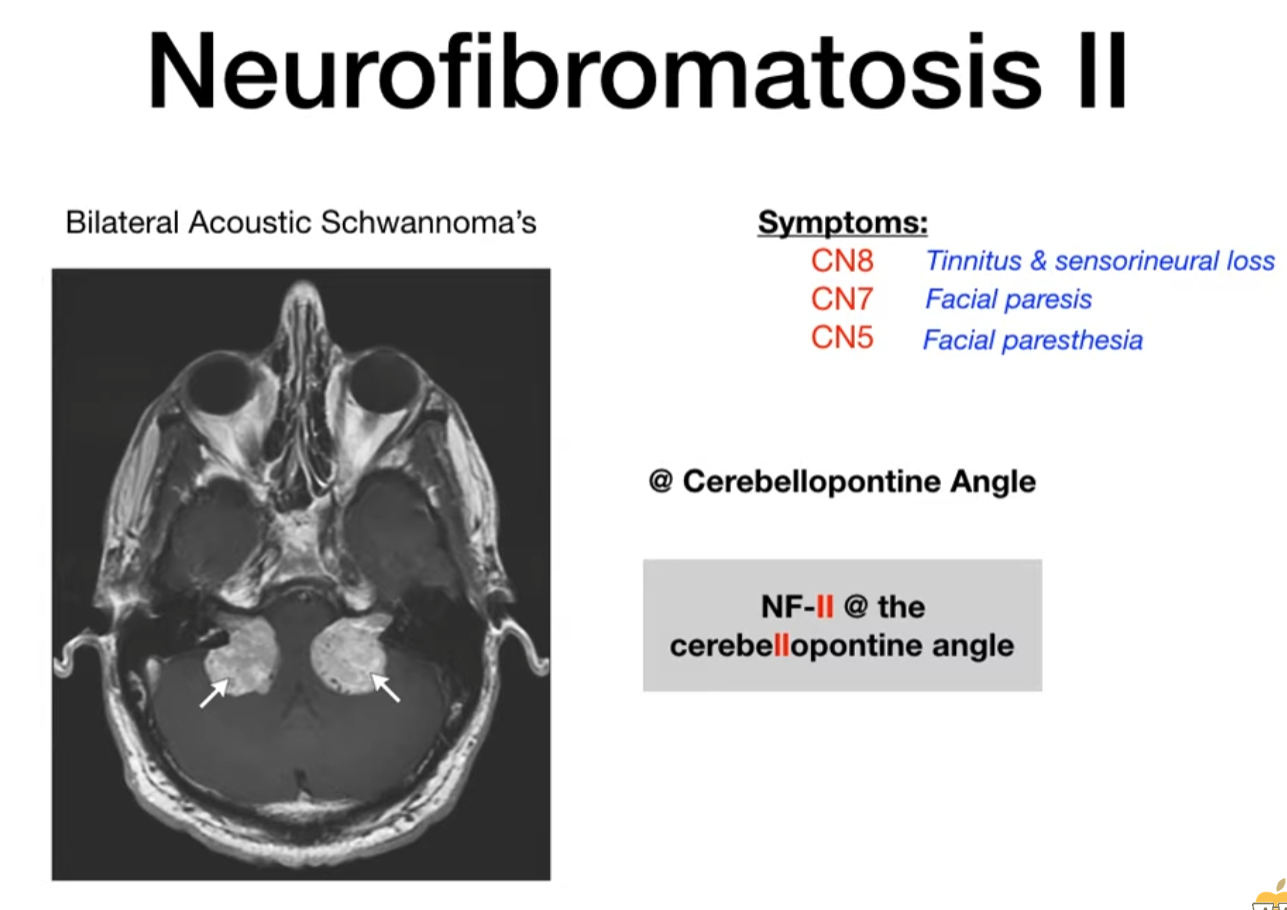
| NF2 | NF2 (Chr 22) | (Fewer/milder skin findings) | Bilateral Vestibular Schwannomas | Multiple meningiomas, Ependymomas |
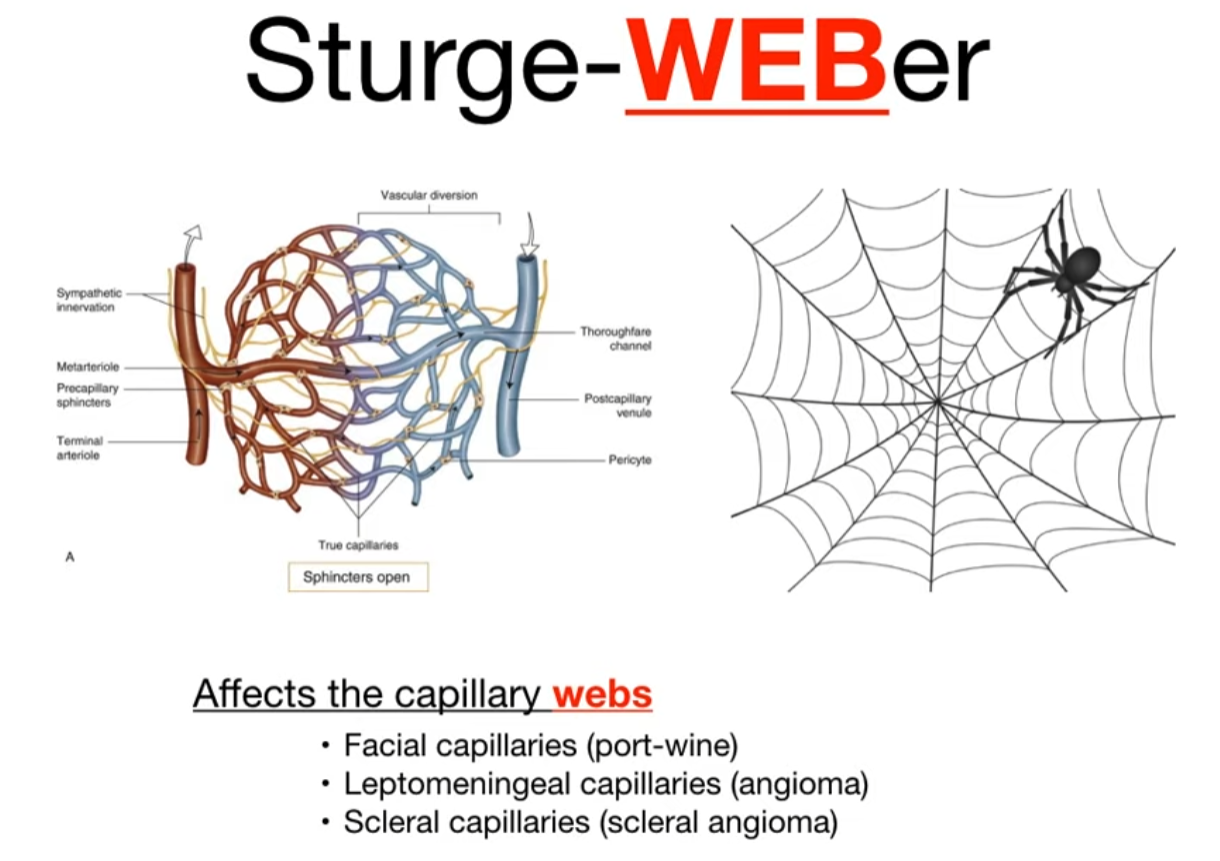
Sturge-Weber syndrome
- Pathophysiology/Etiology
- A rare, sporadic neurocutaneous disorder also known as encephalotrigeminal angiomatosis.
- Caused by a somatic mosaic mutation in the GNAQ gene during early development. This is not an inherited condition.
- The mutation leads to abnormal development and persistence of embryonal blood vessels, resulting in capillary-venous malformations in the skin, brain, and eye.
- Clinical Presentation
- Classic Triad: 1) Facial capillary malformation (port-wine birthmark), 2) Leptomeningeal angioma, and 3) Ocular abnormalities (like glaucoma).
- Skin: A congenital, unilateral port-wine birthmark (nevus flammeus) is the hallmark, typically in the V1 (ophthalmic) distribution of the trigeminal nerve. The likelihood of SWS increases if the V1 distribution is involved.
- Neurologic: Seizures are the most common neurologic feature, often focal and starting in the first year of life. Other findings include developmental delay, intellectual disability, hemiparesis, and headaches.
- Ocular: Glaucoma is a major concern (occurring in 30-70% of patients) and can lead to optic nerve damage and blindness. Other findings include choroidal hemangiomas and buphthalmos (enlargement of the eyeball).
Tuberous sclerosis
- Mutation of tumor suppressor genes (variable expression) → loss of function → unchecked cell growth → tumor development
- Tumor suppressor genes
- TSC1 gene on chromosome 9 encodes hamartin protein
- TSC2 gene on chromosome 16 encodes tuberin protein
- Clinical features
- Intellectual disability (caused by brain lesions)
- Seizures
- Skin manifestations
- Adenoma sebaceum (facial angiofibroma): benign tumor composed of blood vessels and fibrous connective tissue
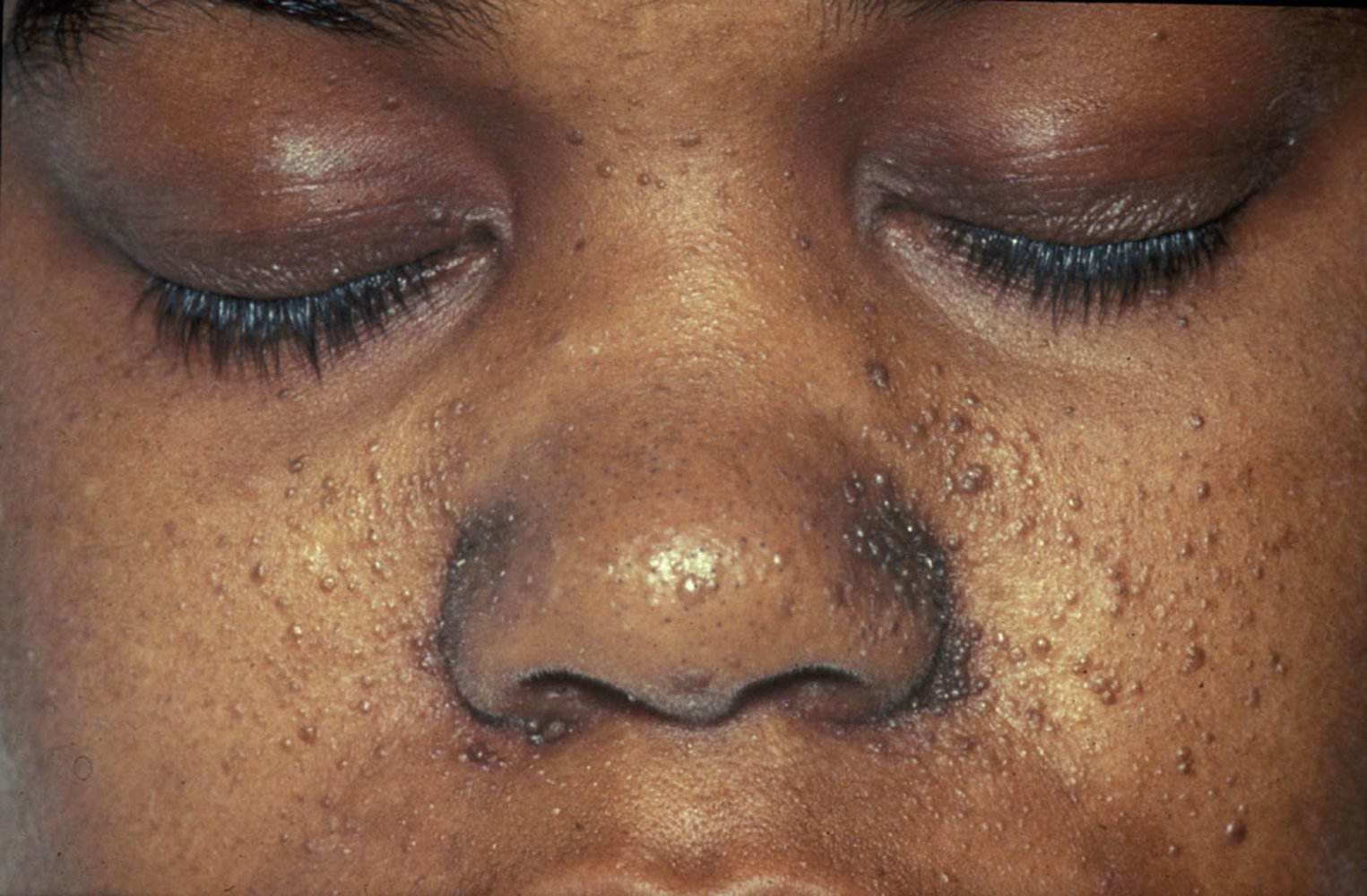
- Mostly located around the nose and cheeks
- Distribution resembles a butterfly shape
- Ash-leaf spots: hypopigmented (white) macules on the trunk and extremities
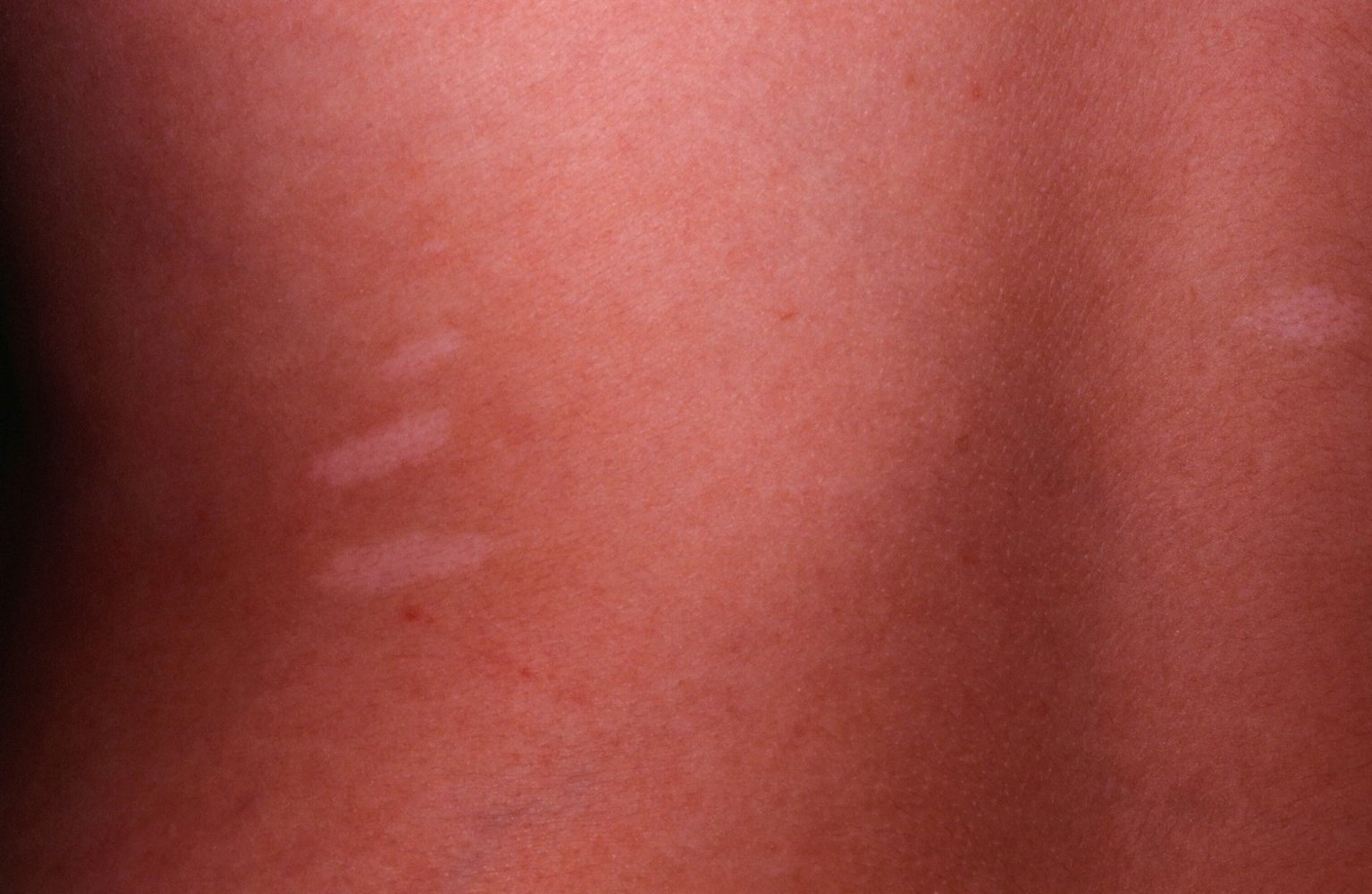
- Shagreen patch: flesh-colored papule in the lumbosacral region with an orange-peel appearance
- Adenoma sebaceum (facial angiofibroma): benign tumor composed of blood vessels and fibrous connective tissue

von Hippel-Lindau syndrome
- Patho/Etiology
- Autosomal dominant mutation of the VHL tumor suppressor gene on chromosome 3p.
- Loss of VHL protein function leads to impaired ubiquitination and elimination of hypoxia-inducible factor 1α (HIF-1α).
- Constitutive upregulation of HIF-1α promotes the transcription of genes for angiogenesis (e.g., VEGF, PDGF) and erythropoietin, leading to the formation of highly vascular tumors.
- Clinical Presentation
- Multisystem disorder characterized by the development of numerous benign and malignant tumors and cysts.
- Onset typically in young adulthood (average age 26).
- Common manifestations include:
- Hemangioblastomas: Most common tumors. Found in the CNS (especially cerebellum and spine) and retina. Cerebellar lesions can cause ataxia, and headaches.
- Renal Cell Carcinoma (RCC): Specifically clear cell subtype, often bilateral and multifocal. Occurs in up to 70% of patients and is the leading cause of mortality. t
- Not renal hemangioma
- Pheochromocytomas: Can be bilateral and cause episodic or sustained hypertension.
- Pancreatic Lesions: Include simple cysts, serous cystadenomas, and neuroendocrine tumors (pNETs).
- Endolymphatic Sac Tumors: Located in the inner ear, can cause hearing loss.
- Epididymal and Broad Ligament Cystadenomas.
- See Hereditary cancer syndromes

Neurofibromatosis
- Etiology
- Neurofibromatosis type 1 and type 2: autosomal dominant inheritance or spontaneous mutation t
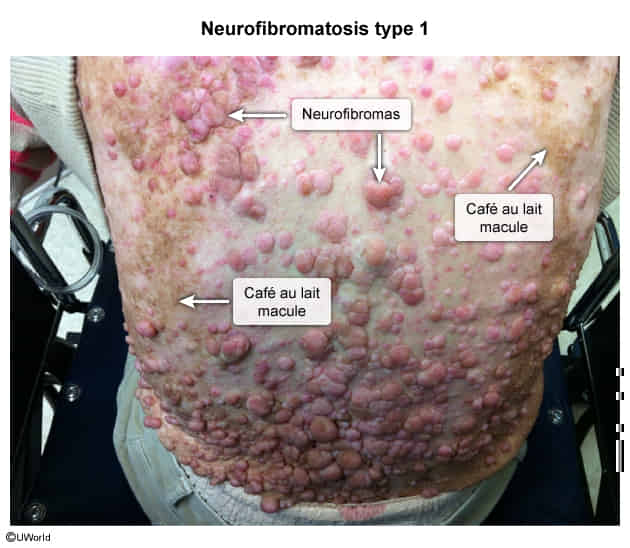
Neurofibromatosis type I
- Café-au-lait spots
Mnemonic
If you drink too much coffee, you are gonna go number one.
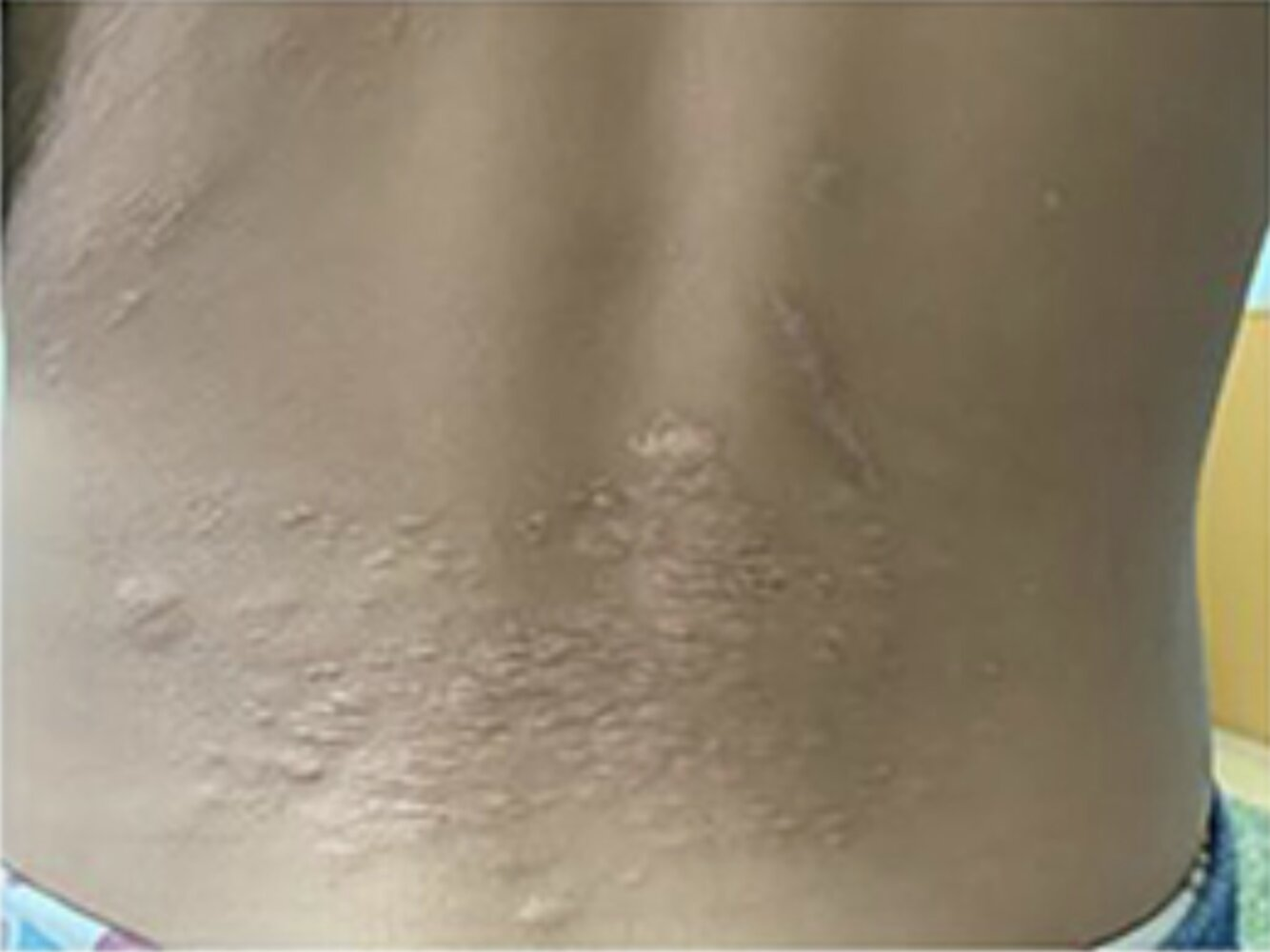
- Cutaneous neurofibromas
- Cutaneous neurofibromas are benign nerve sheath tumors composed of diverse cells and are often found within the dermis. The dermis is composed of collagen, elastic fibers, and ground substance and contains extracellular components including nerves, vasculature, hair follicles, and glands. Neurofibromas are composed of a mixture of cells normally found in peripheral nerves, including neoplastic Schwann cells, as well as non-neoplastic fibroblasts, perineural cells, and mast cells.
- Cutaneous neurofibromas are benign nerve sheath tumors composed of diverse cells and are often found within the dermis. The dermis is composed of collagen, elastic fibers, and ground substance and contains extracellular components including nerves, vasculature, hair follicles, and glands. Neurofibromas are composed of a mixture of cells normally found in peripheral nerves, including neoplastic Schwann cells, as well as non-neoplastic fibroblasts, perineural cells, and mast cells.
- Optic gliomas
Neurofibromatosis type II