Blistering diseases differential diagnostics
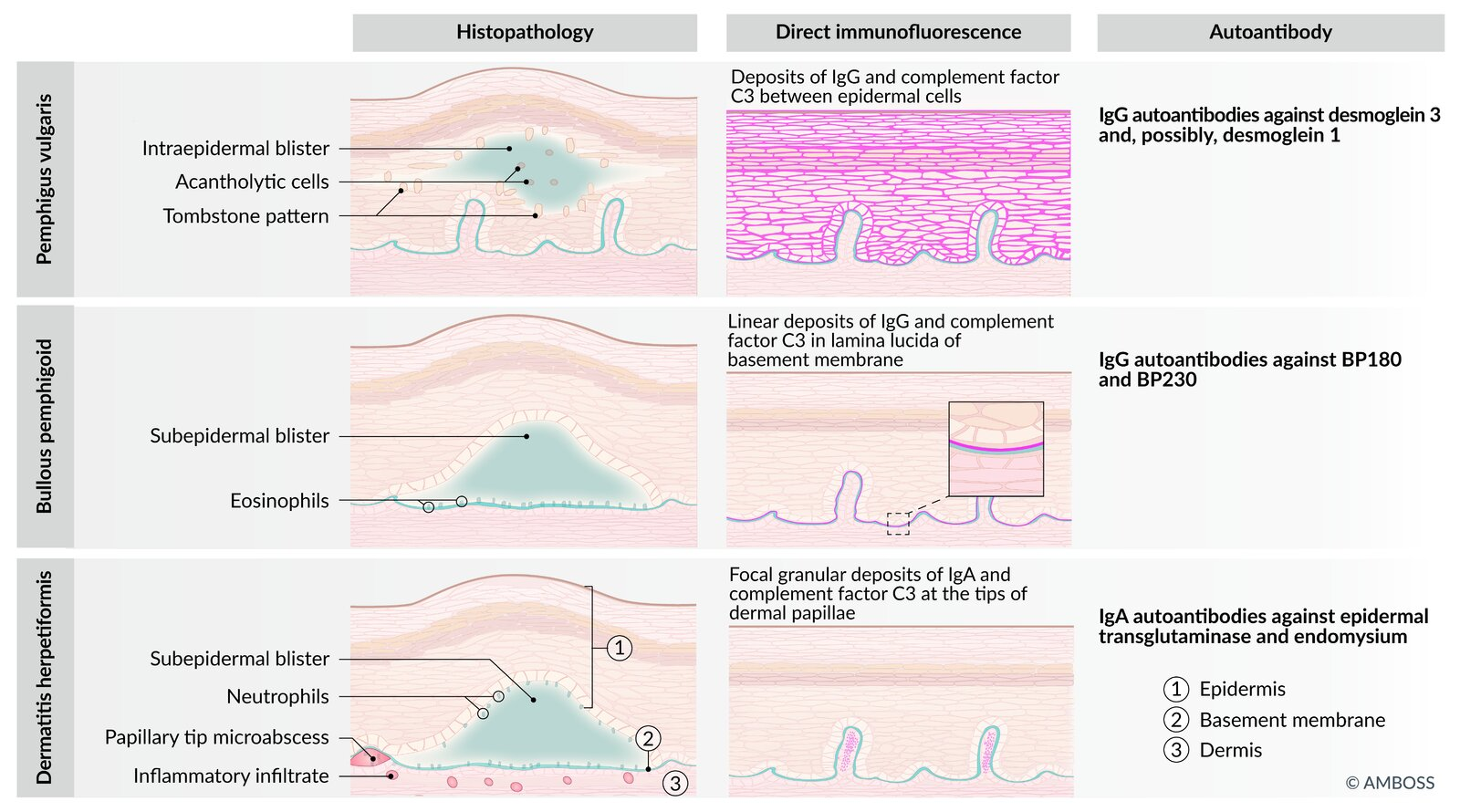
Disease Pathophysiology / Target Antigen Clinical Features & Morphology Histology / Immunofluorescence (IF) Pemphigus Vulgaris IgG against Desmoglein 1 & 3 (Desmosomes)(Type II H.S.) Flaccid bullae; rupture easily.Oral mucosa involved (often 1st).Nikolsky (+) .Intraepidermal split.”Row of tombstones” on basalis.Net-like / Reticular IgG pattern.Bullous Pemphigoid IgG against Hemidesmosomes (BP180/BP230)(Type II H.S.) Tense bullae; do not rupture easily.Nikolsky (-) .Subepidermal split.Eosinophils Linear IgG at basement membrane.Dermatitis Herpetiformis IgA against Tissue Transglutaminase (Cross-reacts w/ reticulin) Pruritic papules/vesicles on extensor surfaces (elbows, knees).Celiac Disease Microabscesses (neutrophils) at dermal papillae tips.Granular IgA at dermal papillae.Epidermolysis Bullosa Hereditary defect in anchoring proteins.(e.g., Keratin 5/14 or Collagen VII) Blisters induced by minor trauma/friction .Presents in infancy/childhood. Cleavage at Dermal-Epidermal Junction (DEJ). Bullous Impetigo Exfoliative Toxin A (S. aureus) cleaves Desmoglein 1 .Flaccid bullae with honey-colored crust .Nikolsky (+) .Subcorneal split (very superficial).Gram (+) cocci in clusters .
Condition Target/Cause Split Location Mucosal? Nikolsky? Key Buzzword SJS/TEN Drugs DEJ (Full necrosis) YES (+) Sloughing skin, >30% BSA (TEN) SSSS S. aureus ToxinGranulosum (Superficial) NO (+) Newborns, Desmoglein-1 split Pemphigus Vulgaris Anti-Desmoglein Suprabasal YES (+) Reticular IF, Tombstone, Flaccid Bullous Pemphigoid Anti-Hemidesmosome Subepidermal No/Rare (-) Linear IF, Tense Bullae Dermatitis Herp. Gluten (IgA) Dermal Papillae No N/A Pruritic, Extensors, Celiac
Link to original
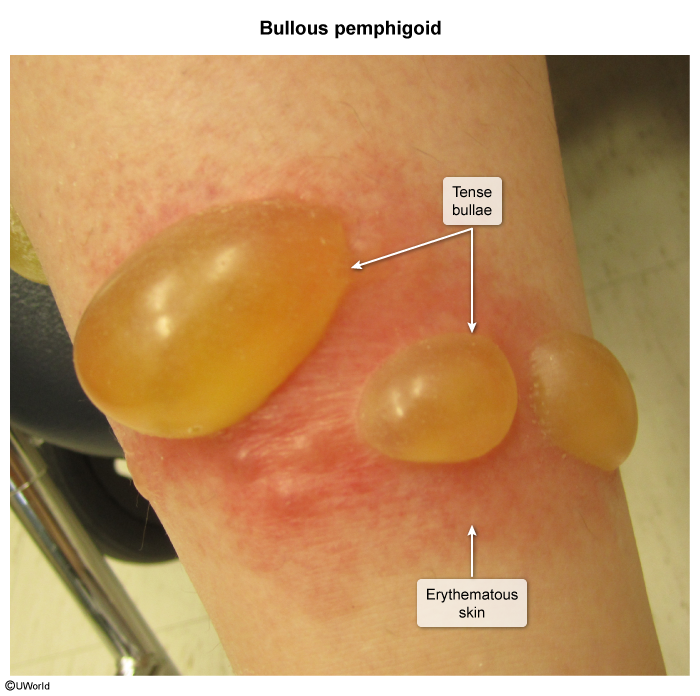
Bullous pemphigoid
Etiology
Type II hypersensitivity reaction
Antihemidesmosome antibodies (IgG)
Clinical findings
Large, tense, subepidermal blisters on normal, erythematous, or erosive skinIntensely pruritic lesions , possibly hemorrhagic, heal without scar formationDistributed on palms, soles, lower legs, groin, and axillae
Oral involvement is rare
Diagnostics
Tzanck test, Nikolsky sign: negative
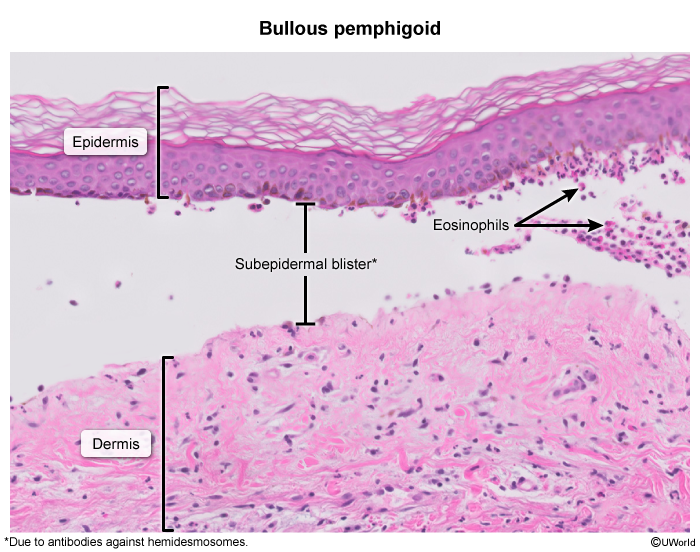
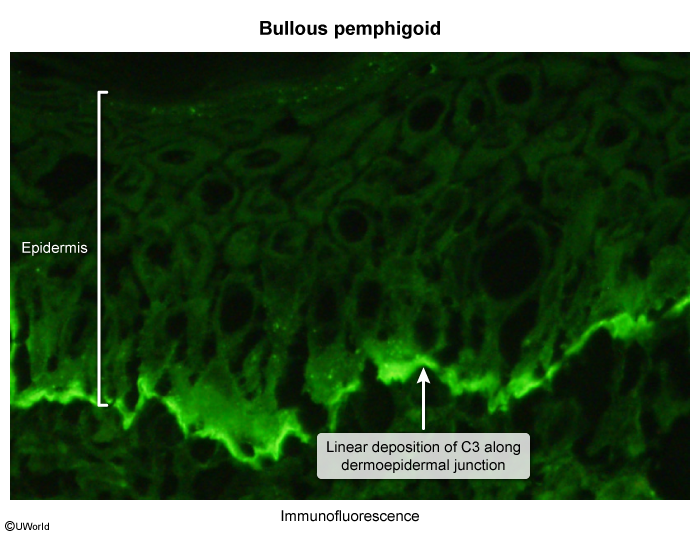
Histology and immunohistochemistry
Subepidermal vesicle formation Eosinophil -rich infiltrate in underlying dermisImmunofluorescence: deposition of linear IgG and C3 along the dermo-epidermal junction
Prognosis
Benign disease , usually responds well to treatment
Blisters are deeper and more robust
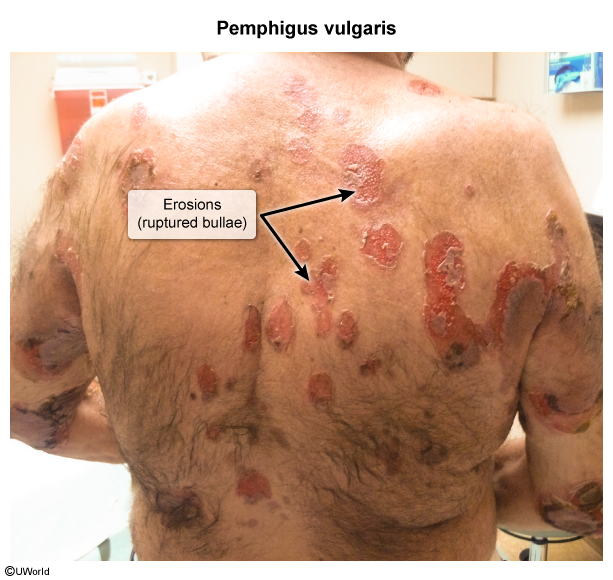
Pemphigus vulgaris
Etiology
Type II hypersensitivity reaction
IgG antibodies directed against desmoglein 3 and desmoglein 1 in desmosome
Clinical findings
Progression in stages
Spontaneous onset of painful flaccid, intraepidermal blisters
Lesions rupture and become confluent → erosions and crusts → re-epithelialization with hyperpigmentation but without scarring
Pruritus is typically absent. Lesions typically first present on the oral mucosa (> 50% of cases), then on body parts exposed to pressure (e.g., intertriginous areas)
Diagnostics
Autoantibodies against
Desmoglein 3 and desmoglein 1
Tzanck test, Nikolsky sign: positive
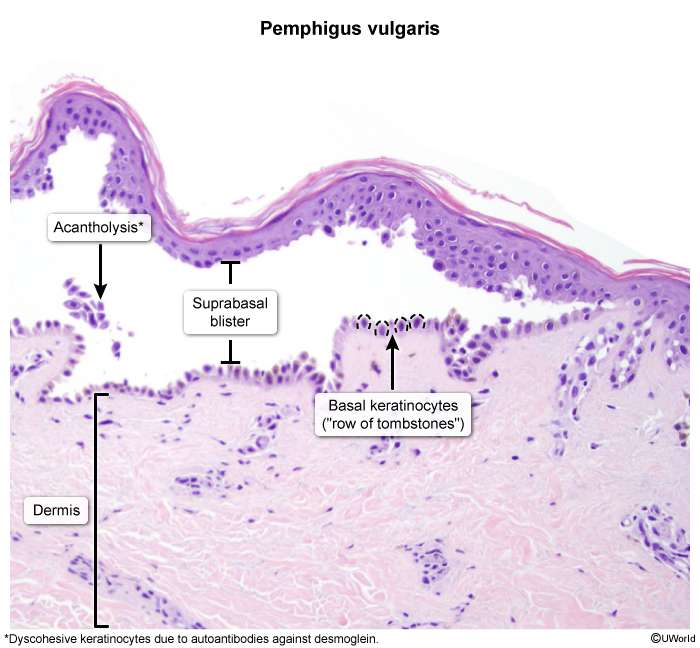
Histology and immunohistochemistry
Intraepidermal vesicle formation just above the basal layer of the epidermis Acantholysis on biopsy: loss of intercellular connections between keratinocytes (“row of tombstones” appearance) Deposition of IgG in the intercellular spaces of the epidermis (esp. early lesions) Immunofluorescence: deposition of IgG in a reticular pattern around epidermal cells
Prognosis
Often fatal without treatment!
Caused primarily by infections, fluid loss, and electrolyte disturbances
Fragile blisters rupture easily
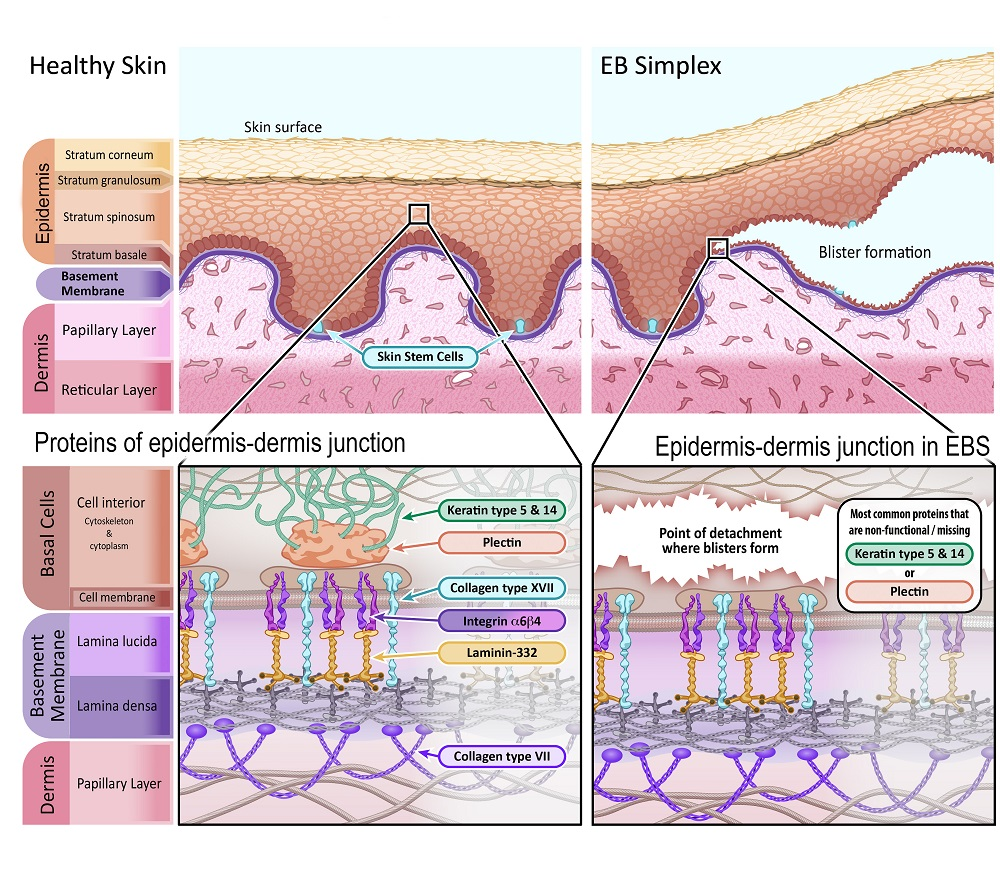
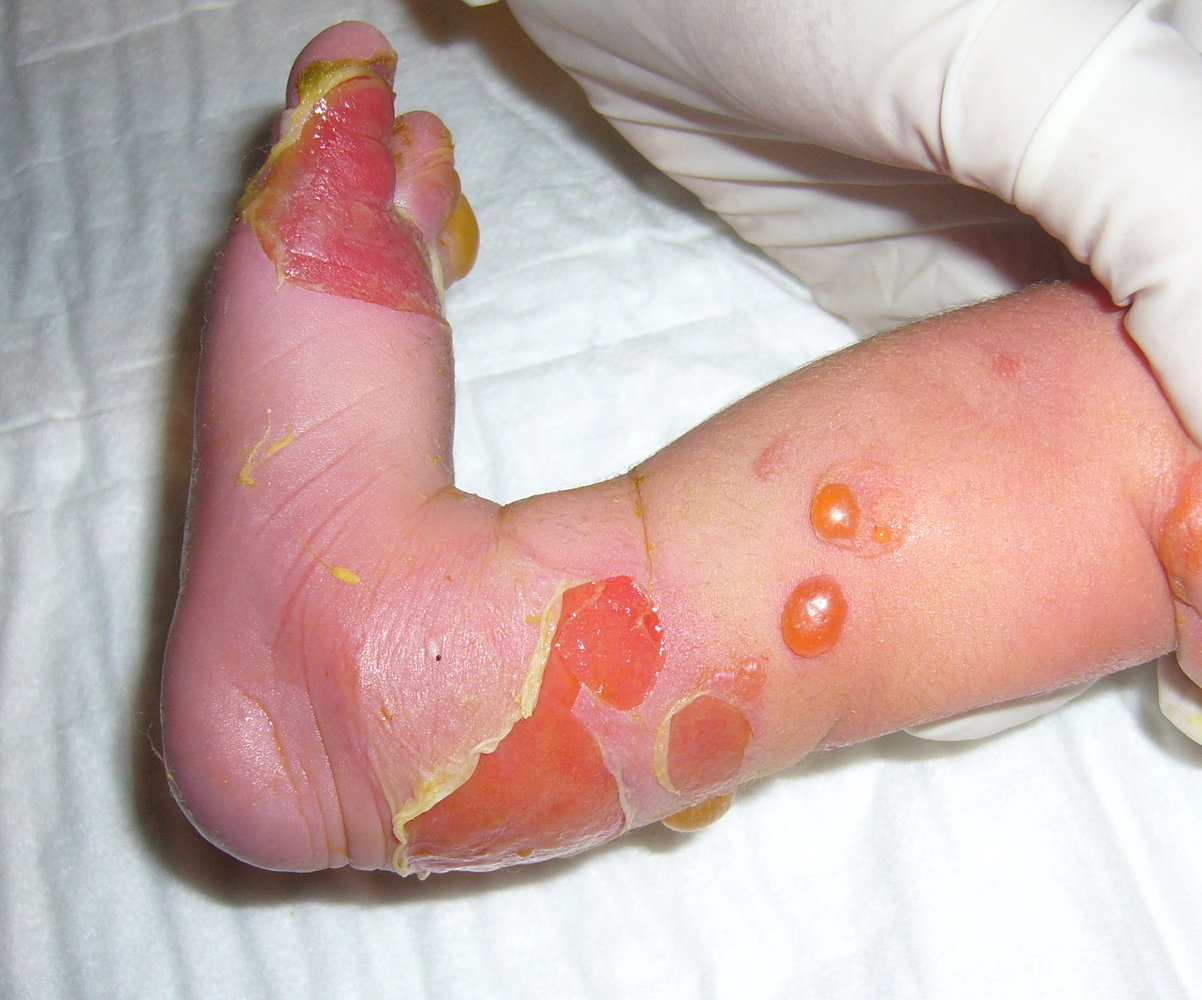
Epidermolysis bullosa
Definition: a genetic condition that causes the skin to become very fragile and blister easily in response to minor injury or friction Epidemiology: EBS is the most common type of EB.
Etiology
Pathophysiology: mutations in keratin proteins → defective assembly of keratin filaments → disruption of the basal layer of keratinocytes → ↑ fragility of epithelial tissue
Clinical features
Mainly limited to the palms and soles Generally heal without scarring