A neurodegenerative disease with upper and lower motor neuron dysfunction. Pure motor neuron disease
Epidemiology
- Mean age of onset is 65 years.
Etiology
Pathophysiology
- Neurodegenerative disorder affecting both Upper Motor Neurons (UMN) and Lower Motor Neurons (LMN).
- Most cases are sporadic (90-95%).
- Familial cases (5-10%) often associated with superoxide dismutase 1 (SOD1) mutation (zinc-copper superoxide dismutase mutation).
- Mechanism: Oxidative stress, glutamate excitotoxicity, and protein aggregation lead to neuronal death.
- Degeneration of:
- Anterior horn cells of spinal cord (LMN signs).
- Corticospinal tract (UMN signs).
- Motor nuclei of brainstem (CN V, IX, X, XII).
- Focal Onset:
- The degenerative process in ALS does not begin in all motor neurons simultaneously. It starts in a specific, focal group of motor neurons
- Pattern of Spread (Prion-like Propagation):
- The leading hypothesis is that the pathogenic proteins (e.g., misfolded TDP-43) propagate from sick neurons to healthy, synaptically connected neurons.
Clinical features
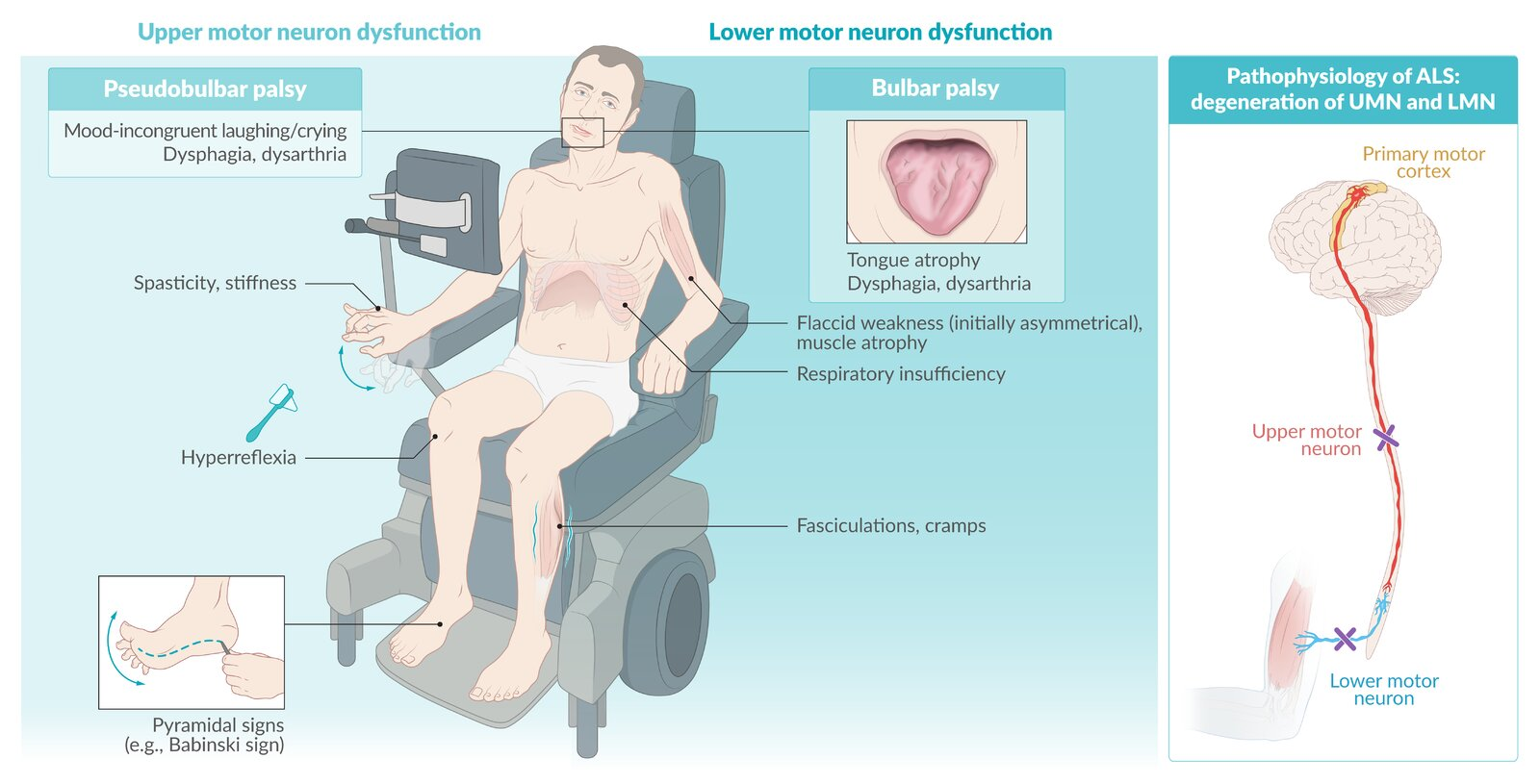
General disease characteristics
- Both upper motor neuron (UMN) and lower motor neuron (LMN) signs are present (see Upper motor neuron (UMN) injury vs. lower motor neuron (LMN) injury)
- Constant disease progression: it usually starts in one arm and/or leg then progresses to the contralateral side
Early symptoms
- Symptoms are highly variable and potentially non-specific (e.g., subtle vocal changes or difficulties grasping objects)
- Asymmetric limb weakness, often beginning with weakness in the hands and feet
- Bulbar symptoms such as dysarthria, dysphagia, and tongue atrophy (20% of cases at disease onset)
- Pseudobulbar palsy with pseudobulbar affect may develop.
- Fasciculations, cramps, and muscle stiffness
- Weight loss
- Split hand sign: a wasting pattern in which the muscles of the thenar eminence atrophy due to degeneration of the lateral portion of the anterior horn of the spinal cord
Late symptoms
- Cognitive impairment (approx. 15% of ALS patients meet the criteria for frontotemporal dementia)
- Autonomic symptoms (e.g., constipation, bladder dysfunction) may develop; the mechanism of development is unclear.
- Life-threatening symptoms
- Respiratory failure due to paralysis of respiratory muscles
- Dysphagia due to bulbar weakness or pseudobulbar palsy
Diagnosis
- Electromyography
- Denervation: fibrillations, positive sharp waves, and large amplitudes
- Fasciculations
Differential Diagnosis
- Myasthenia gravis
- Weakness improves with acetylcholinesterase inhibitors
- No UMN or LMN signs
Treatment
- Riluzole
- A sodium-channel blocker that inhibits glutamate release in the CNS and decreases glutamate excitotoxicity
- Prolongs survival and slows functional decline in patients with ALS (on average, for 3 months)
- Edaravone
- A free radical scavenger
- Has been shown to slow functional decline in some patients with ALS