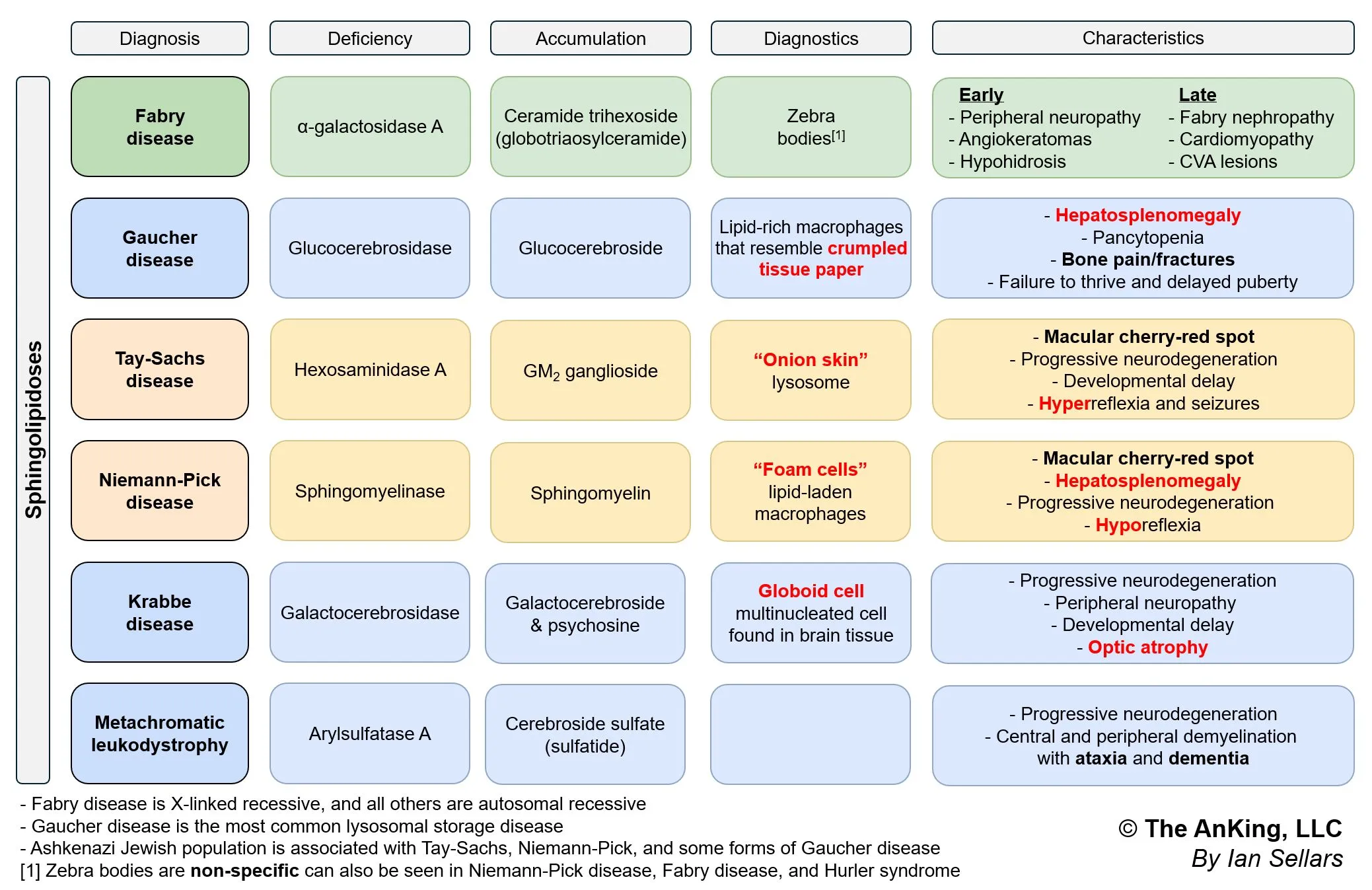
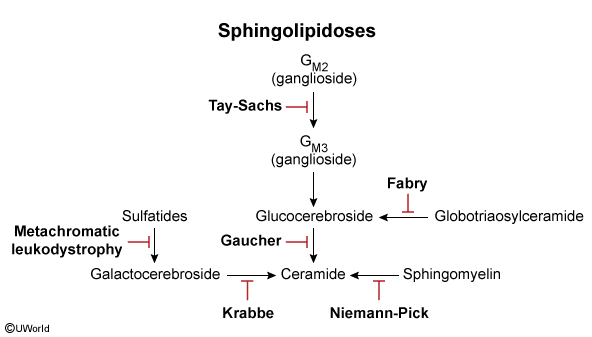
- Tay-Sachs Disease: “Cherry-red” macula, NO hepatosplenomegaly, hyperreflexia.
- Niemann-Pick Disease: “Cherry-red” macula, WITH hepatosplenomegaly, foam cells.
- Gaucher Disease: Hepatosplenomegaly, pancytopenia, bone crises, “wrinkled tissue paper” macrophages.
- Fabry Disease: Peripheral neuropathy (burning pain), angiokeratomas, hypohidrosis, X-linked.
- Krabbe Disease: Peripheral neuropathy, optic atrophy, developmental delay, globoid cells.
- Metachromatic Leukodystrophy: Central & peripheral demyelination, ataxia, dementia.
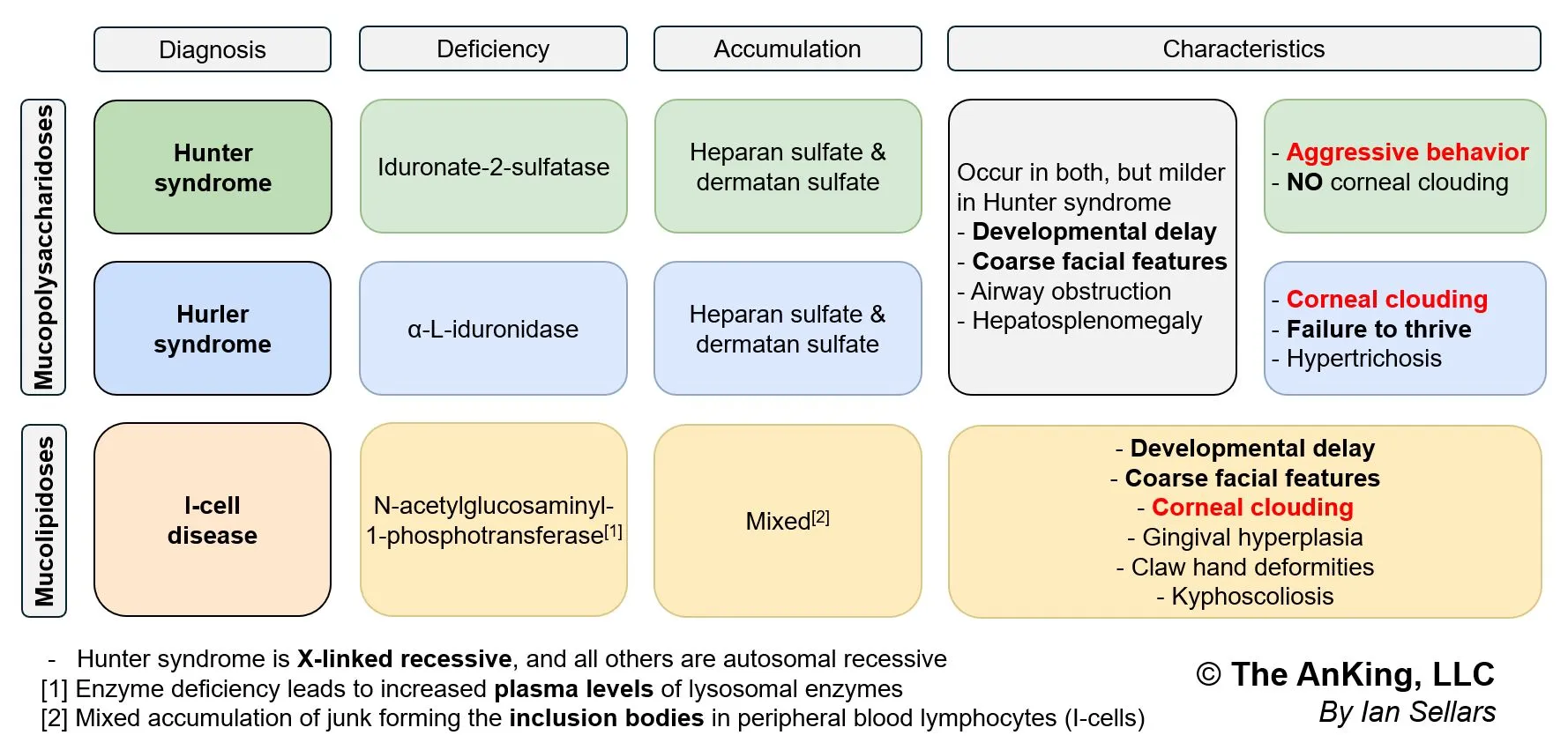
- Hurler Syndrome (MPS I): Corneal clouding, gargoylism, developmental delay.
- Hunter Syndrome (MPS II): NO corneal clouding, aggressive behavior, X-linked.
- I-cell disease: Coarse facial features, gingival hyperplasia, corneal clouding, restricted joint movements, high plasma levels of lysosomal enzymes.
Fabry disease
 Fabrite → favourite
Fabrite → favourite
- Features (Classic Triad):
-
- Episodic peripheral neuropathy (burning pain in hands/feet).
-
- Angiokeratomas (dark red skin lesions).
-
- Hypohidrosis (decreased sweating).
-
- Late Complications: Renal failure, cardiovascular disease.
Gaucher disease
 Because you are crying, you need a tissue paper to wipe tears.
Because you are crying, you need a tissue paper to wipe tears.
- Features:
- Most common LSD.
- Massive hepatosplenomegaly.
- Pancytopenia (anemia, thrombocytopenia, etc.).
- Aseptic necrosis of femur, bone crises.
- Gaucher cells: Lipid-laden macrophages resembling “crinkled tissue paper”.
Tay-Sachs disease
 Epidemiology: more common in the Ashkenazi Jewish population
small = no hepatosplenomegaly
Epidemiology: more common in the Ashkenazi Jewish population
small = no hepatosplenomegaly
Mnemonic
- Any diseases with a hyphen in the name will presents with Cherry-Red Macula.
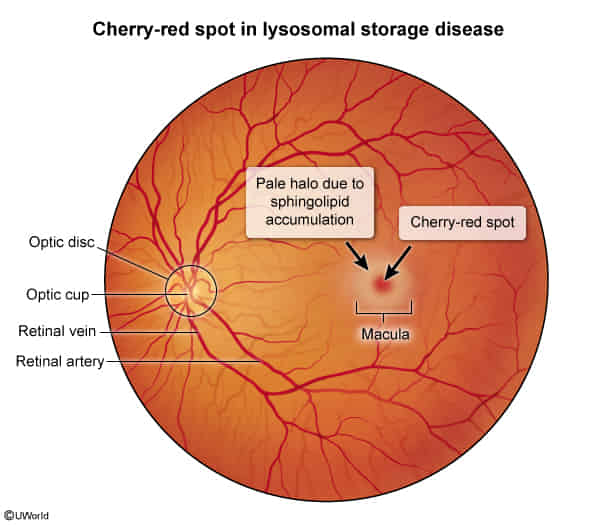
- Retinal Ganglion Cell Accumulation: In these diseases, the defective lysosomal enzymes lead to an accumulation of undigested substrates (like lipids) within the cells, including the ganglion cells of the retina.
- Macular Opacity: The ganglion cell layer is particularly thick in the macula, the central part of the retina responsible for sharp, detailed vision. The buildup of these materials in the ganglion cells around the fovea makes this area appear pale or opaque.
- Foveal Contrast: The fovea centralis, the very center of the macula, is extremely thin and lacks ganglion cells. It derives its blood supply from the choroid, not the retinal circulation. Therefore, it’s not affected by the storage material accumulation in the same way.
- Cherry-Red Appearance: Because the surrounding area becomes pale due to the accumulated substances, the unaffected fovea maintains its normal reddish appearance (due to the underlying choroidal blood supply), creating a contrasting “cherry-red spot” in the center.
Niemann-Pick disease

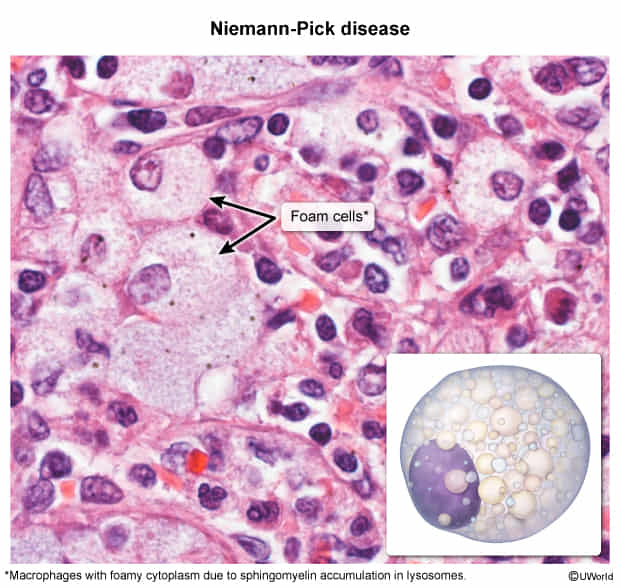 Sphinger → finger
big = hepatosplenomegaly
Sphinger → finger
big = hepatosplenomegaly
Krabbe disease
 World → Galaxy → Gala
World → Galaxy → Gala
Hunter syndrome & Hurler syndrome

Mnemonic
- X → X link recessive
- The gun touches the hunter’s derm → Heparan sulfate, dermatan sulfate.
- Hunter doesn’t have corneal cloudy → or he couldn’t see targets
- Hunters are aggressive
- Hurler syndrome
- Hunter syndrome
- Milder features than Hurler:
- (+) Aggressive behavior
- (−) Corneal clouding
- Death in adolescence/early adulthood
- Milder features than Hurler:
Metachromatic leukodystrophy

- Metapod can’t walk, can’t say, can demyelinate to butterfly
Tip
Pompe disease also affects lysosome.
I-cell disease
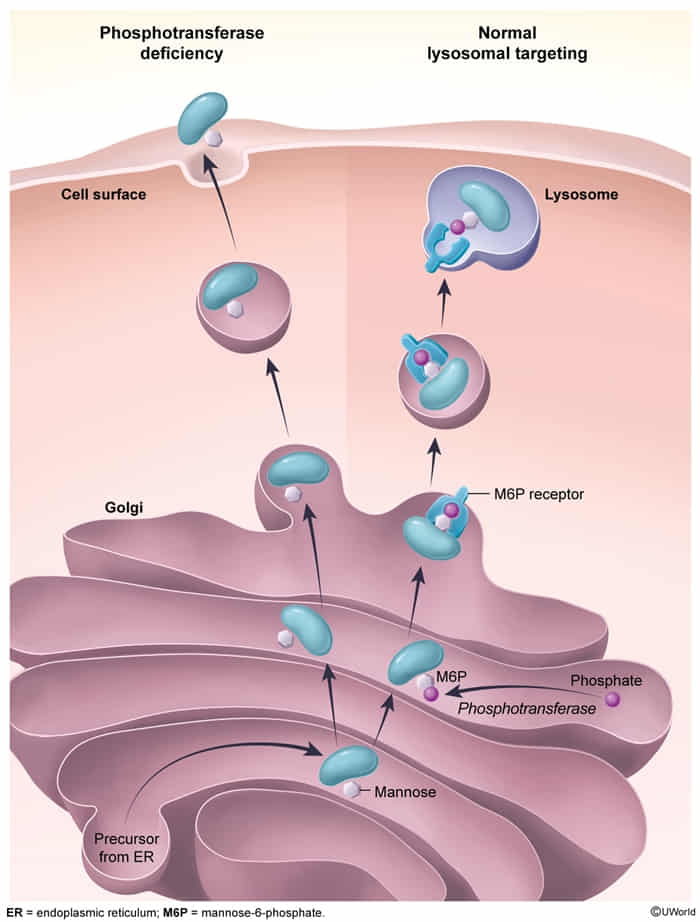
- Definition: an autosomal recessive disease caused by a defect in N-acetylglucosaminyl-1-phosphotransferase activity
- Pathophysiology
- This enzyme is responsible for adding the Mannose-6-Phosphate (M6P) tag to newly synthesized lysosomal enzymes in the Golgi apparatus.
- Without the M6P tag, lysosomal enzymes are not sorted correctly to the lysosome.
- Instead, these enzymes are secreted extracellularly.
- Result: Lysosomes lack necessary enzymes → accumulation of undigested substrates (glycosaminoglycans and lipids) within the lysosome → formation of Inclusion bodies.
- Clinical features
- Presentation typically in infancy (failure to thrive).
- Coarse facial features.
- Corneal clouding.
- Restricted joint movement and claw hand deformities.
- Skeletal abnormalities (Dysostosis multiplex, kyphoscoliosis).
- Gingival hyperplasia.
- Severe developmental delay/intellectual disability.
- Note: Symptoms resemble Hurler syndrome (Mucopolysaccharidosis I) but are more severe and appear earlier.
- Diagnostics
- Serum Labs: ↑ Lysosomal enzymes in the plasma (extracellularly) is the diagnostic hallmark.
- Histology: Fibroblasts show dense intracytoplasmic inclusions (“I-cells”).
- Imaging: Skeletal survey showing dysostosis multiplex.