- Von Gierke (Type I): Glucose-6-phosphatase deficiency impairs gluconeogenesis and glycogenolysis, causing severe fasting hypoglycemia, hepatomegaly, and lactic acidosis.
- Pompe (Type II): Lysosomal acid maltase deficiency leads to glycogen accumulation in lysosomes, resulting in fatal cardiomyopathy (“Pompe trashes the Pump”) and hypotonia.
- Cori (Type III): Debranching enzyme deficiency results in the accumulation of limit dextrins and a milder hypoglycemia than Type I with normal lactate levels.
- McArdle (Type V): Myophosphorylase deficiency in skeletal muscle causes painful muscle cramps and myoglobinuria with exercise, characterized by a flat venous lactate curve.
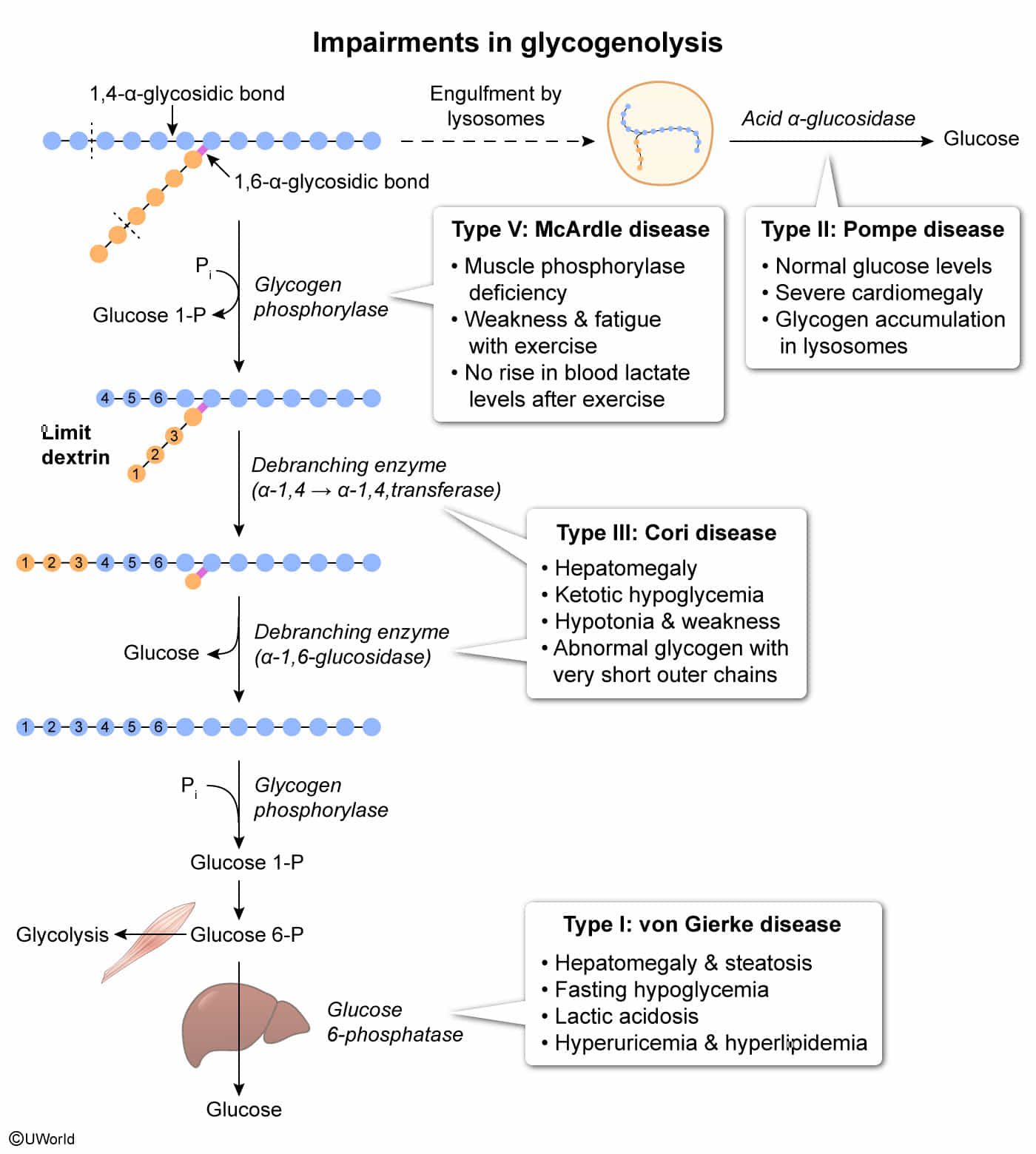
Glycogen metabolism
- Common Step: Glycogen Glucose-1-P Glucose-6-P (G6P).
- Most glycogen breakdown occurs in the cytosol via glycogen phosphorylase, but a small, clinically important fraction (~1–3%) is degraded in lysosomes. This pathway mainly serves glycogen “housekeeping,” clearing abnormal or excess glycogen rather than supplying energy. Therefore, Pompe disease will have normal glucose level.
- Liver (Maintains Blood Sugar)
- Has Glucose-6-Phosphatase.
- G6P Free Glucose Bloodstream.
- Muscle (Fuel for Contraction)
- LACKS Glucose-6-Phosphatase.
- G6P is trapped inside the cell.
- G6P Glycolysis Pyruvate + ATP.
Type I: Von Gierke Disease
- Pathophysiology/Etiology: Deficient Glucose-6-Phosphatase. This impairs both glycogenolysis and gluconeogenesis, preventing the liver from releasing free glucose into the blood.
- Clinical Presentation: Presents in infancy (3-6 months) with a doll-like face, thin extremities, and a protuberant abdomen (due to massive hepatomegaly).
- Severe fasting hypoglycemia is a hallmark, often leading to seizures.
- Diagnosis:
- Labs: ↑ Lactate (lactic acidosis), ↑ uric acid (gout), ↑ triglycerides/hyperlipidemia.
- Glucagon challenge test: Blood glucose fails to rise.
- Management: Frequent oral glucose/cornstarch feedings, avoidance of fructose and galactose.
Type II: Pompe Disease
- Pathophysiology/Etiology: Deficient lysosomal α-1,4-glucosidase (acid maltase) t . Glycogen accumulates in lysosomes, leading to cellular damage. Pompe Pumps the heart.
- Pompe Disease is the ONLY Glycogen Storage Disease that is also a Lysosomal Storage Disease.
- Clinical Presentation:
- Classic Infantile form: Presents in early infancy with cardiomegaly (often hypertrophic cardiomyopathy), profound muscular hypotonia (“floppy baby”), feeding difficulties, and macroglossia. Death often occurs from cardiorespiratory failure.
- Normal blood glucose levels.
- Diagnosis:
- Enzyme assay showing deficient acid maltase activity.
- PAS stain shows glycogen accumulation within lysosomes.
- ↑ CK and LDH levels.
- Management: Enzyme replacement therapy.
Type III: Cori Disease
- Pathophysiology/Etiology: Deficient debranching enzyme (α-1,6-glucosidase). Glycogenolysis is incomplete, resulting in the accumulation of limit dextrin-like structures.
- Clinical Presentation: A milder version of Von Gierke disease.
- Presents with hepatomegaly, hypoglycemia, and short stature.
- Gluconeogenesis is intact, so lactic acid and uric acid levels are usually normal.
- Skeletal and cardiac muscle involvement can occur.
- Diagnosis:
- Normal blood lactate levels distinguish it from Von Gierke.
- Biopsy shows glycogen with abnormally short outer branches.
- Management: High-protein diet to support gluconeogenesis.
Type V: McArdle Disease
- Pathophysiology/Etiology: Deficient skeletal muscle glycogen phosphorylase (myophosphorylase). This prevents the breakdown of glycogen in muscles for energy. McArdle = Muscle.
- Clinical Presentation: Presents in adolescence or early adulthood.
- Exercise intolerance, painful muscle cramps, and fatigue, especially with strenuous activity.
- Myoglobinuria (reddish-brown urine) after exercise.
- “Second wind” phenomenon: Symptoms improve with rest or continued exercise at a lower intensity as blood flow increases, delivering glucose and fatty acids.
- Diagnosis:
- Flat venous lactate curve during forearm exercise test (no rise in lactate), with a normal rise in ammonia.
- Blood glucose levels are typically unaffected.
- ↑ CK during episodes.
- Management: Avoidance of strenuous exercise. Consuming simple carbohydrates before exercise can be beneficial.
Von Gierke disease

- Hepatomegaly and Renomegaly:
- Glucose-6-phosphate cannot be converted to glucose, so it accumulates in liver and kidney cells
- This excess G6P gets converted to glycogen, which builds up in these organs
- The chronic accumulation causes organ enlargement and can lead to hepatic adenomas over time
- The other symptoms occur because the body can’t access glucose properly:
Cori disease
 Abnormal glycogen branch like coral.
Abnormal glycogen branch like coral.
- Clinical presentation is similar to that of type I, though symptoms tend to be less severe.
- Generalized muscle weakness and/or cramps
- Possibly cirrhosis (ascites, hepatomegaly, and splenomegaly)
- Mild, fasting hypoglycemia and ketosis
- Hyperlipidemia
- Cytosolic accumulations of limit dextrin-like complexes
- Blood lactate levels are normal.
- Gluconeogenesis is functional.
- Type III may result in cardiomyopathy.
McArdle disease & Hers disease

Mnemonic
Muscle → McArdle’s Hepatic → Her’s
- Flat venous lactate curve with exaggerated elevations in blood ammonia during exercise
- No glucose, can’t produce lactate
- Increased protein and amino acid Metabolism
Anderson disease

Mnemonic
Mr. Anderson’s black coat is long and straight.

Pompe disease

- This condition is caused by deficiency of acid alpha-glucosidase (alpha-1,4 glucosidase or acid maltase), an enzyme responsible for breaking down glycogen within the acidic environment of lysosomes.
- Although most glycogen is degraded in the cytoplasm, a small percentage is inadvertently engulfed by lysosomes, especially in cells containing high amounts of glycogen such as hepatocytes and myocytes. Deficiency of acid maltase results in pathologic accumulation of glycogen within liver and muscle lysosomes. Cardiac and skeletal muscle are particularly susceptible because the ballooning lysosomes interfere with contractile function.