- Location: Mitochondria.
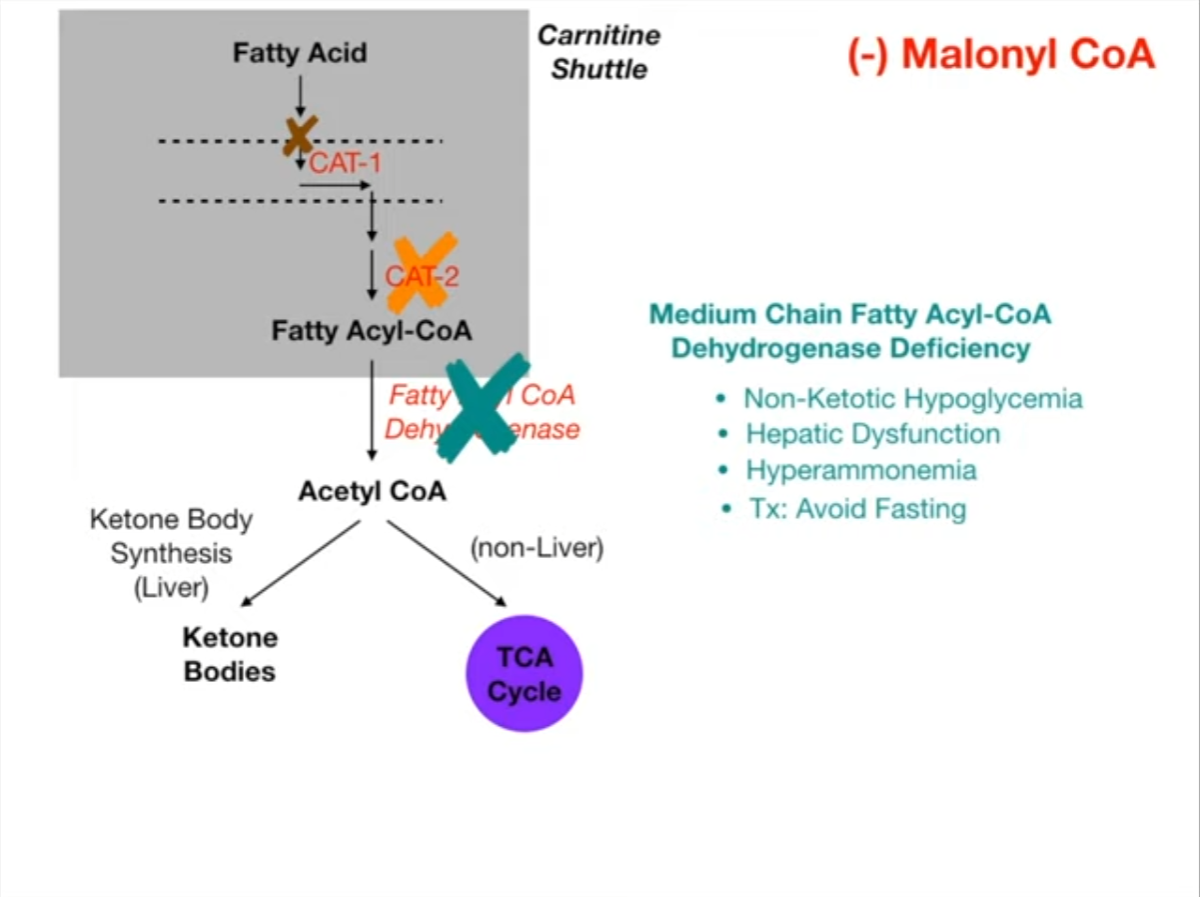
- Rate-Limiting Enzyme: Carnitine Palmitoyltransferase I (CPT I).
- Regulation:
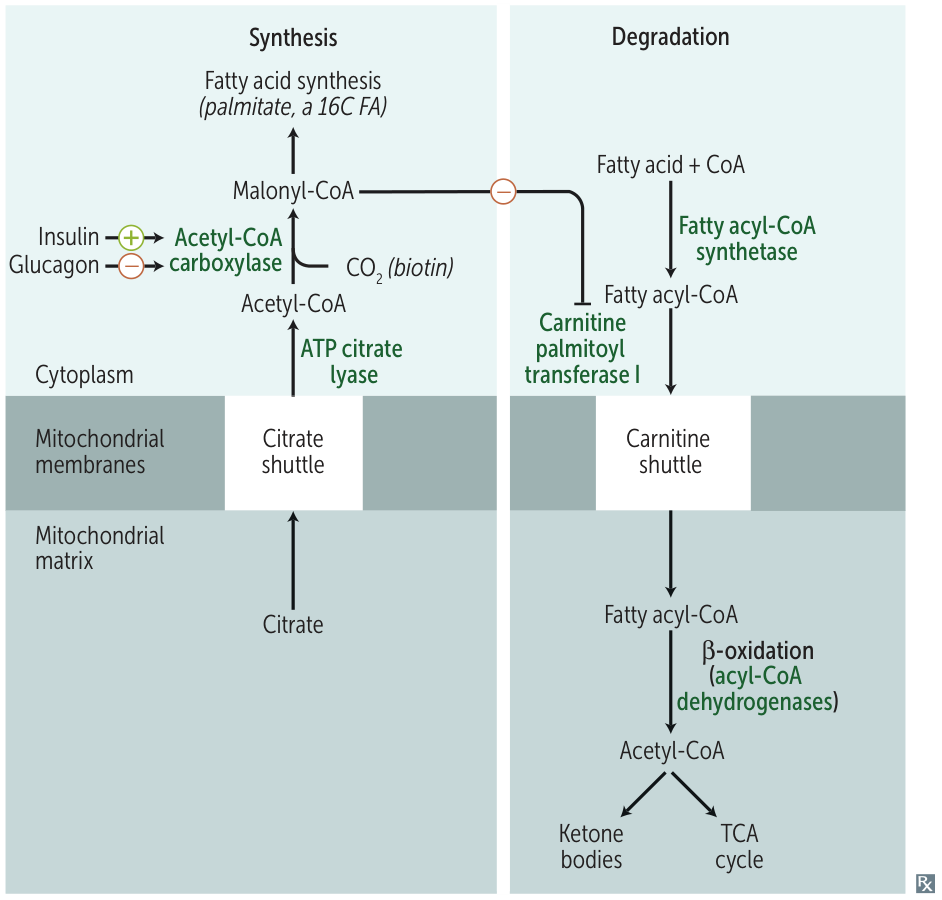
- Inhibited by: Malonyl-CoA (prevents simultaneous breakdown & synthesis). t
- Transport: Carnitine Shuttle transports Long-Chain FAs into mitochondrial matrix.
- Products: Acetyl-CoA (→ TCA/Ketones), NADH, FADH2.
- Odd-Chain FAs:
- Yield Propionyl-CoA → Methylmalonyl-CoA → Succinyl-CoA.
- Cofactors: Biotin (B7) and Vitamin B12.
- Clinical Pearl: Only part of FA that contributes to Gluconeogenesis.
- Pathology: MCAD Deficiency
- AR defect in Medium-Chain Acyl-CoA Dehydrogenase.
- Trigger: Fasting/Illness.
- Hallmark: Hypoketotic Hypoglycemia (low glucose, low ketones).
- Note: Differentiates from Type 1 DM DKA (Hyperketotic Hyperglycemia).
Pathophysiology/Etiology
- Function: Breaks down fatty acids (FAs) into acetyl-CoA, generating NADH and FADH2. This is a crucial energy source during fasting, starvation, and prolonged exercise, especially for the heart and skeletal muscle.
- Location: Primarily in the mitochondrial matrix.
- Process Overview: A cyclical process that sequentially cleaves two-carbon units (as acetyl-CoA) from the fatty acyl-CoA molecule.
Key Steps:
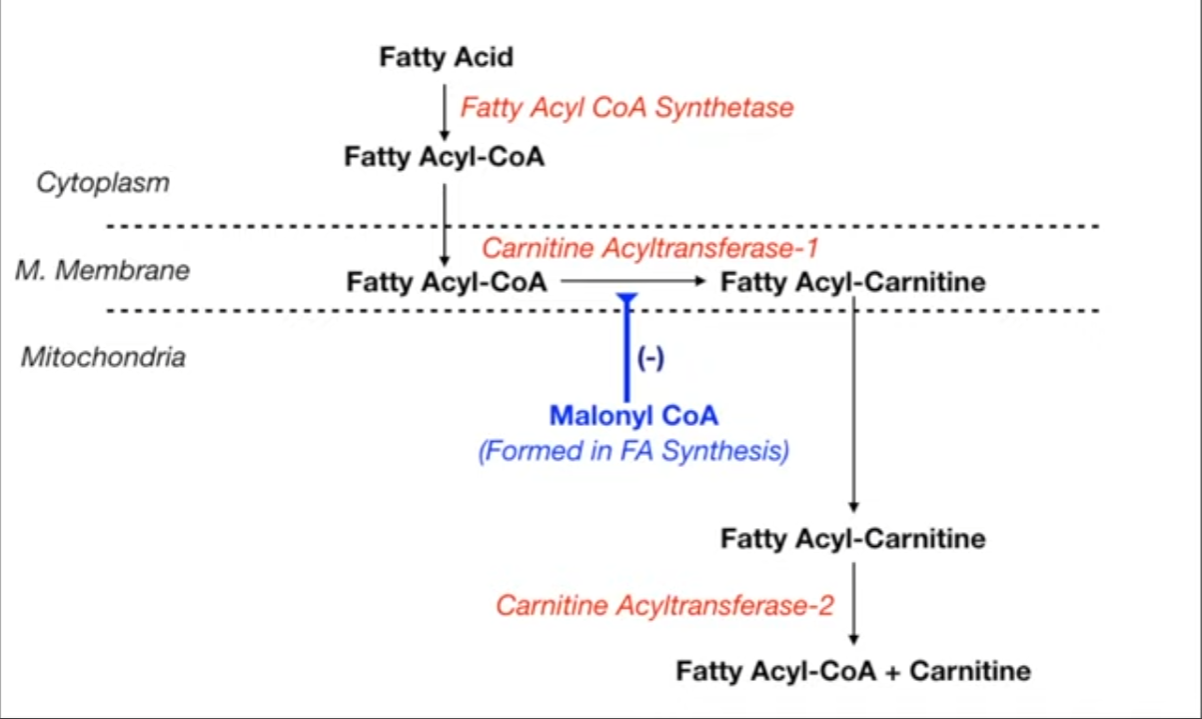
- Activation: In the cytoplasm, long-chain fatty acids (LCFAs) are activated to fatty acyl-CoA by fatty acyl-CoA synthetase, requiring ATP.
- Transport (Rate-Limiting Step): LCFAs require the carnitine shuttle to enter the mitochondria.
- CPT1 (Carnitine Palmitoyltransferase I): Fatty acyl-CoA is converted to acylcarnitine on the outer mitochondrial membrane. Inhibited by Malonyl-CoA (an intermediate in FA synthesis).
- Translocase: Acylcarnitine is transported into the mitochondrial matrix.
- CPT2 (Carnitine Palmitoyltransferase II): Acylcarnitine is converted back to fatty acyl-CoA in the matrix.
- Note: Short and medium-chain FAs do not require the carnitine shuttle.
- β-Oxidation Spiral: In the mitochondrial matrix, a four-reaction sequence is repeated:
- Oxidation by acyl-CoA dehydrogenase (produces FADH2).
- Hydration.
- Oxidation by β-hydroxyacyl-CoA dehydrogenase (produces NADH).
- Thiolysis (cleavage) to release acetyl-CoA and a fatty acyl-CoA that is two carbons shorter.
- Fates of Acetyl-CoA:
- Enters the TCA cycle in muscle and other tissues for ATP production.
- Used for ketone body synthesis in the liver, especially during fasting.
Clinical Presentation of Defects
- Disorders typically present during periods of catabolic stress (e.g., fasting, illness, prolonged exercise).
- Classic presentation: Hypoketotic hypoglycemia. The hypoglycemia occurs because gluconeogenesis requires ATP and NADH, which are supplied by β-oxidation. Without β-oxidation, gluconeogenesis is impaired. Ketone body production is also deficient.
- Other common symptoms include lethargy, vomiting, seizures, coma, hyperammonemia, and myopathy.
Key Disorders
Carnitine Shuttle Defects
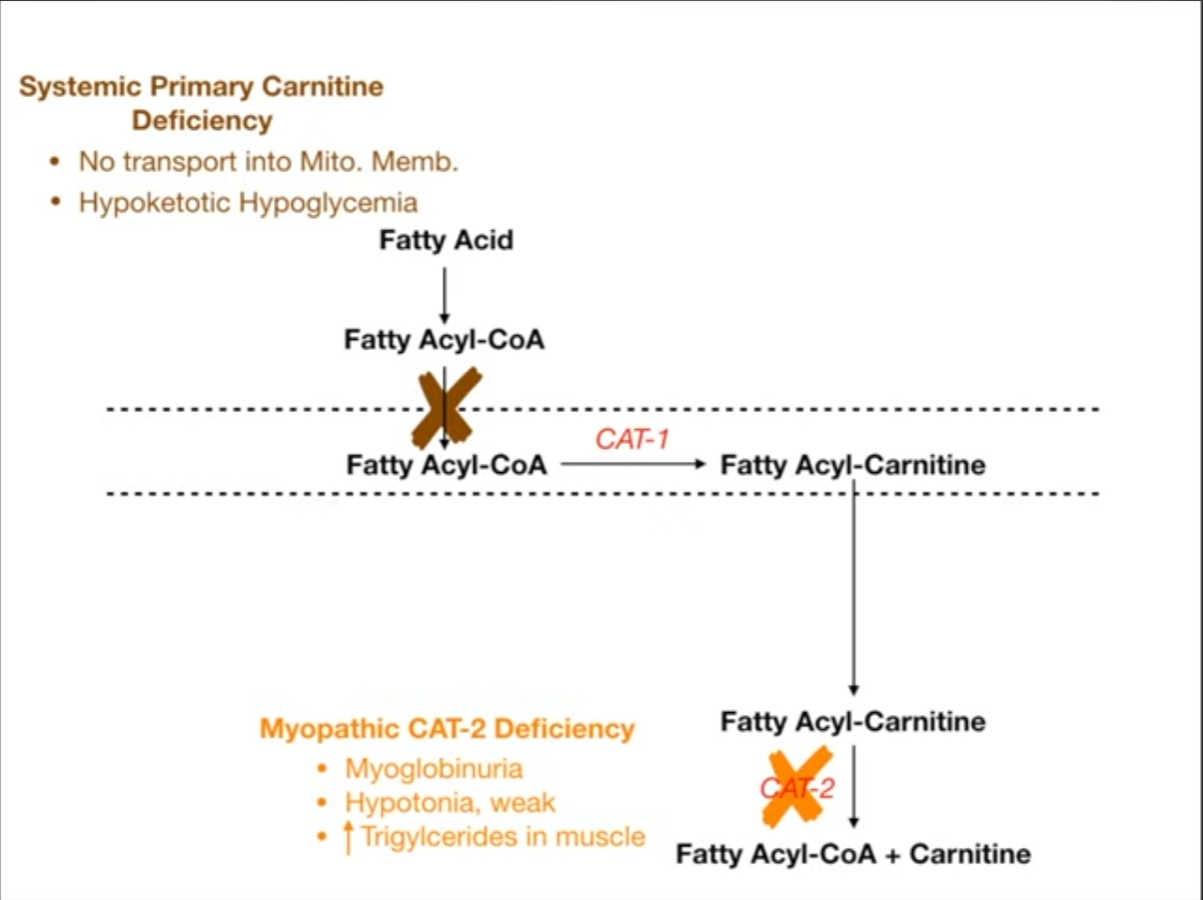
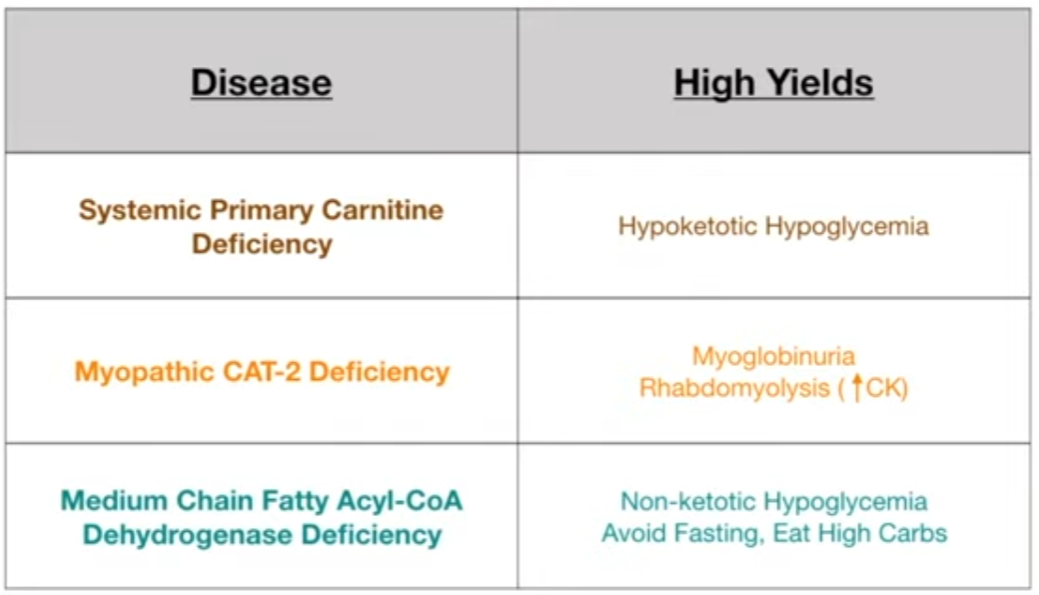
- Primary Carnitine Deficiency: Defect in the carnitine transporter.
- Dx: Low plasma carnitine levels, reduced carnitine uptake by cells.
- Presentation: Weakness, hypotonia, cardiomyopathy, hypoketotic hypoglycemia.
- Carnitine Palmitoyltransferase (CPT) I Deficiency: Affects the liver.
- Dx: Elevated plasma carnitine, acylcarnitine profile shows ↑ free carnitine.
- Presentation: Primarily hepatic symptoms with hypoketotic hypoglycemia and risk of liver failure.
- Carnitine Palmitoyltransferase (CPT) II Deficiency: Most common inherited disorder of lipid metabolism in adults.
- Dx: Acylcarnitine profile shows elevated C16, C18:1 acylcarnitines during an attack.
- Presentation: Classic adult form involves myalgia, muscle weakness, and myoglobinuria (red-brown urine) triggered by prolonged exercise, fasting, or illness.
Acyl-CoA Dehydrogenase Deficiencies
- Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency: Most common inborn error of FA oxidation.
- Dx: Elevated C8-C10 acylcarnitines in plasma, dicarboxylic acids in urine. Now part of newborn screening. t
- Presentation: Presents in infancy or early childhood. Vomiting, lethargy, seizures, coma, liver dysfunction, hypoketotic hypoglycemia following a period of fasting (e.g., viral illness).
- Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency:
- Dx: Elevated C14-C18 acylcarnitines.
- Presentation: Can present with severe cardiomyopathy in infancy, hypoketotic hypoglycemia, or later-onset myopathy.
Management/Treatment
- Acute: IV dextrose to correct hypoglycemia and reverse catabolism.
- Long-term:
- Avoid fasting and prolonged exercise.
- Provide frequent meals with a high-carbohydrate, low-fat diet.
- Carnitine supplementation for carnitine deficiencies.
- Medium-chain triglyceride (MCT) oil can be used as a supplement in LCHAD/VLCAD deficiency, as MCTs bypass the carnitine shuttle.
Key Associations/Buzzwords
- Buzzwords: Hypoketotic hypoglycemia, myoglobinuria after exercise, dicarboxylic aciduria.
- Jamaican Vomiting Sickness: Caused by eating unripe ackee fruit, which contains hypoglycin A. This toxin irreversibly inhibits MCAD, leading to symptoms mimicking MCAD deficiency.
- Odd-Chain Fatty Acids: Undergo β-oxidation to yield acetyl-CoA and one molecule of propionyl-CoA. Propionyl-CoA enters the TCA cycle after being converted to succinyl-CoA.
- Peroxisomal Oxidation: Very-long-chain fatty acids (>20 carbons) t are initially oxidized in peroxisomes. Defects lead to conditions like Zellweger syndrome or X-linked adrenoleukodystrophy.

Rate-limiting enzyme
Tip
Very long chain fatty acids (VLCFAs) and certain branched-chain fatty acids (eg, phytanic acid) cannot undergo mitochondrial beta-oxidation; these fatty acids are metabolized by a special form of beta-oxidation (VLCFAs) or by alpha-oxidation (branched-chain fatty acids) within peroxisomes. t