Summary of steps (important!)
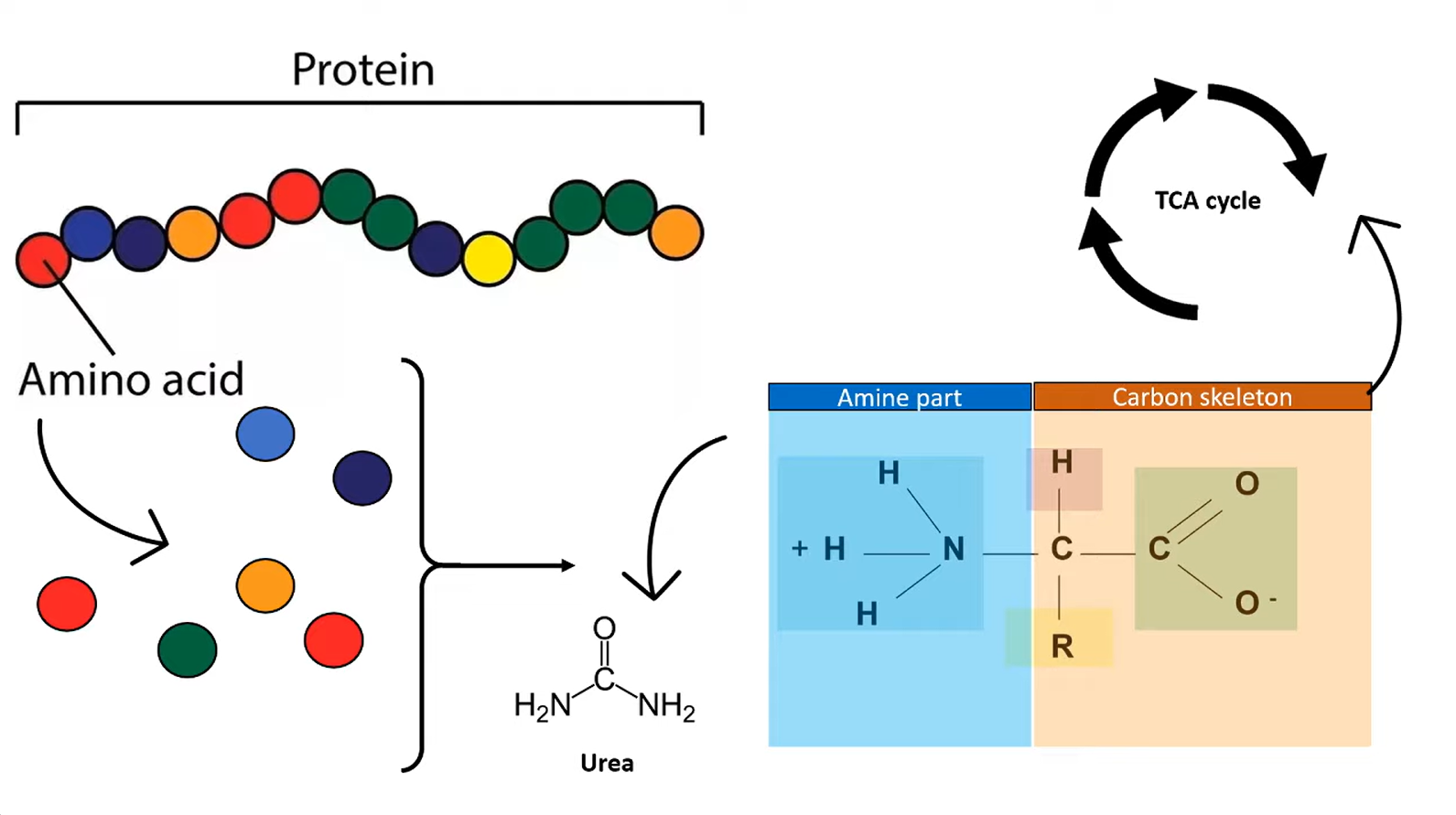
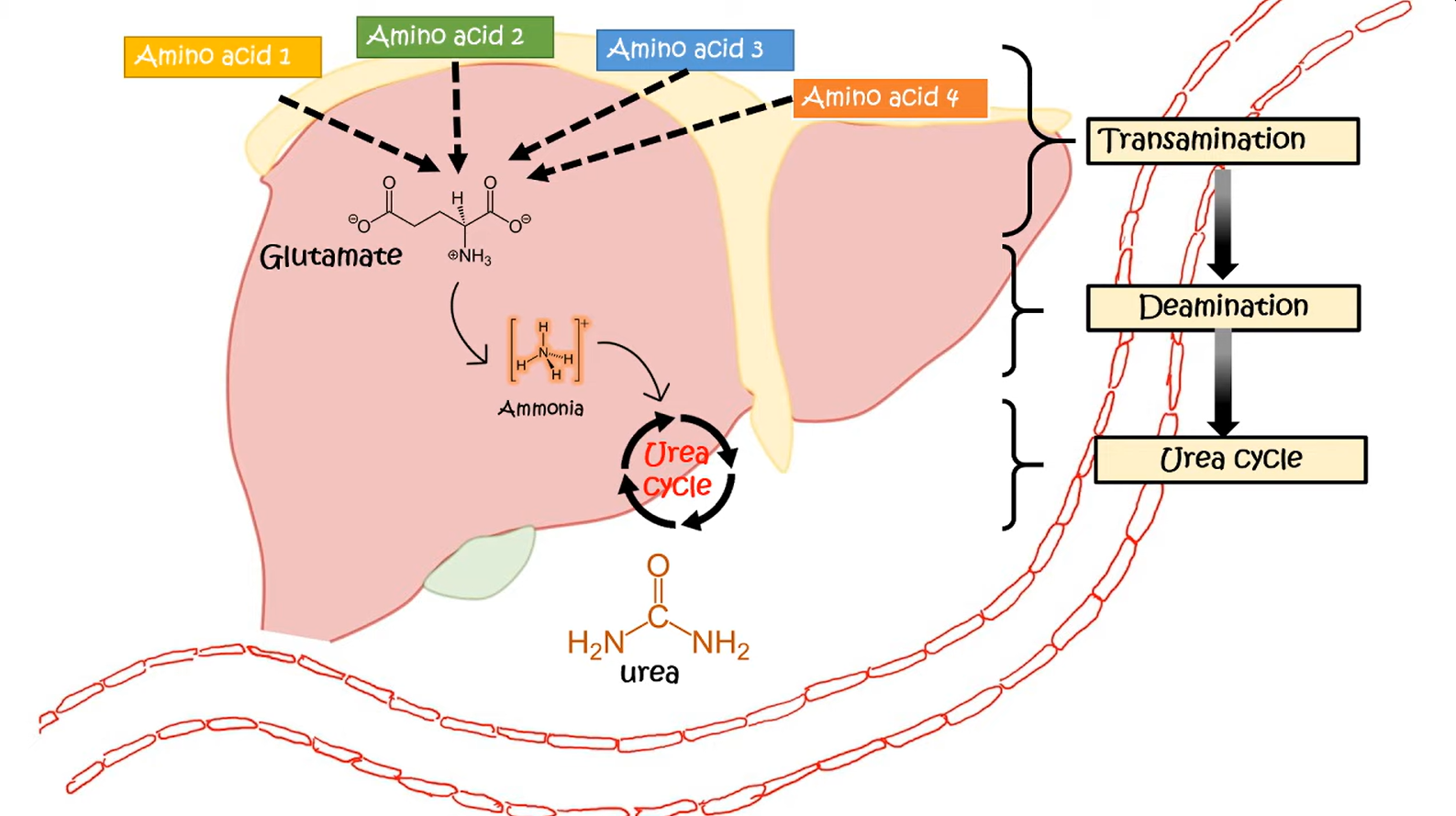
- Generation of ammonia in periphery
- Transportation of ammonia to liver
- In periphery
- Transamination: amino acids + α-ketoglutarate ⇄ α-ketoacids + glutamate
- Transport to liver, by either
- Glutamine cycle (most common)
- Primary Sites: Brain and Peripheral Tissues.
- Purpose: The major mechanism for scavenging excess ammonia.
- Glutamate + NH4+ + ATP → Glutamine + ADP + Pi
- Alanine cycle (Cahill cycle)
- Primary Site: Skeletal Muscle.
- Purpose: Transports nitrogen from muscle protein breakdown to the liver while regenerating glucose for muscle energy.
- Pyruvate + glutamate ⇄ alanine + α-ketoglutarate
- Glutamine cycle (most common)
- In liver
- Convert back to glutamate, by either
- Glutaminase: glutamine + H2O → glutamate + ammonium
- Transamination: alanine + α-ketoglutarate ⇄ pyruvate + glutamate
- Release ammonia (Deamination): glutamate + NAD(P)+ + H2O ⇄ α-ketoglutarate + NH4+ + NAD(P)H + H+
- Convert back to glutamate, by either
- In periphery
- Excretion of ammonia
- Urea cycle
| Feature | Glutamate | Glutamine |
|---|---|---|
| Primary Role | Major excitatory neurotransmitter in CNS | Nitrogen transport; fuel for gut & immune cells |
| Structure | Acidic (anionic); side chain has -COOH | Neutral; side chain has -CONH2 (amide) |
| Metabolism | Made from glutamine (via glutaminase) | Made from glutamate (via glutamine synthetase) |
| Key Assoc. | Excitotoxicity (e.g., stroke, seizure) | Conditionally essential (in stress/illness) |
| CNS Cycle | Released by neurons → taken up by glia | Released by glia → taken up by neurons |
Transamination
- Description: transfer of an amino group from an AA to an α-ketoacid for breakdown, or to an α-ketoacid to form a nonessential AA
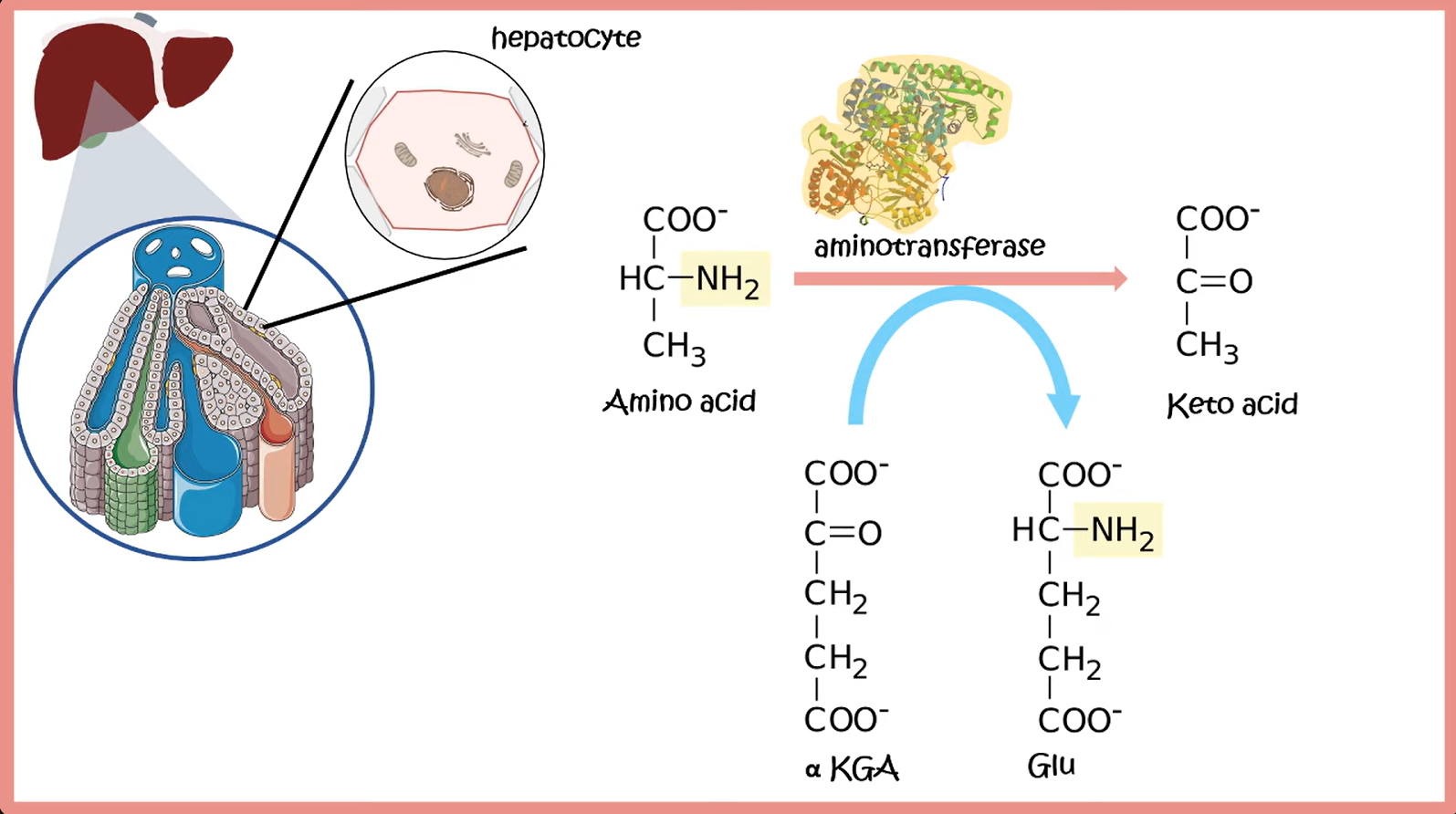
- E.g.
- ALT:
- AST: aspartate + α-ketoglutarate ⇄ oxalacetate + glutamate
Deamination
- Description: reaction in which an amino group from an AA is released as ammonium
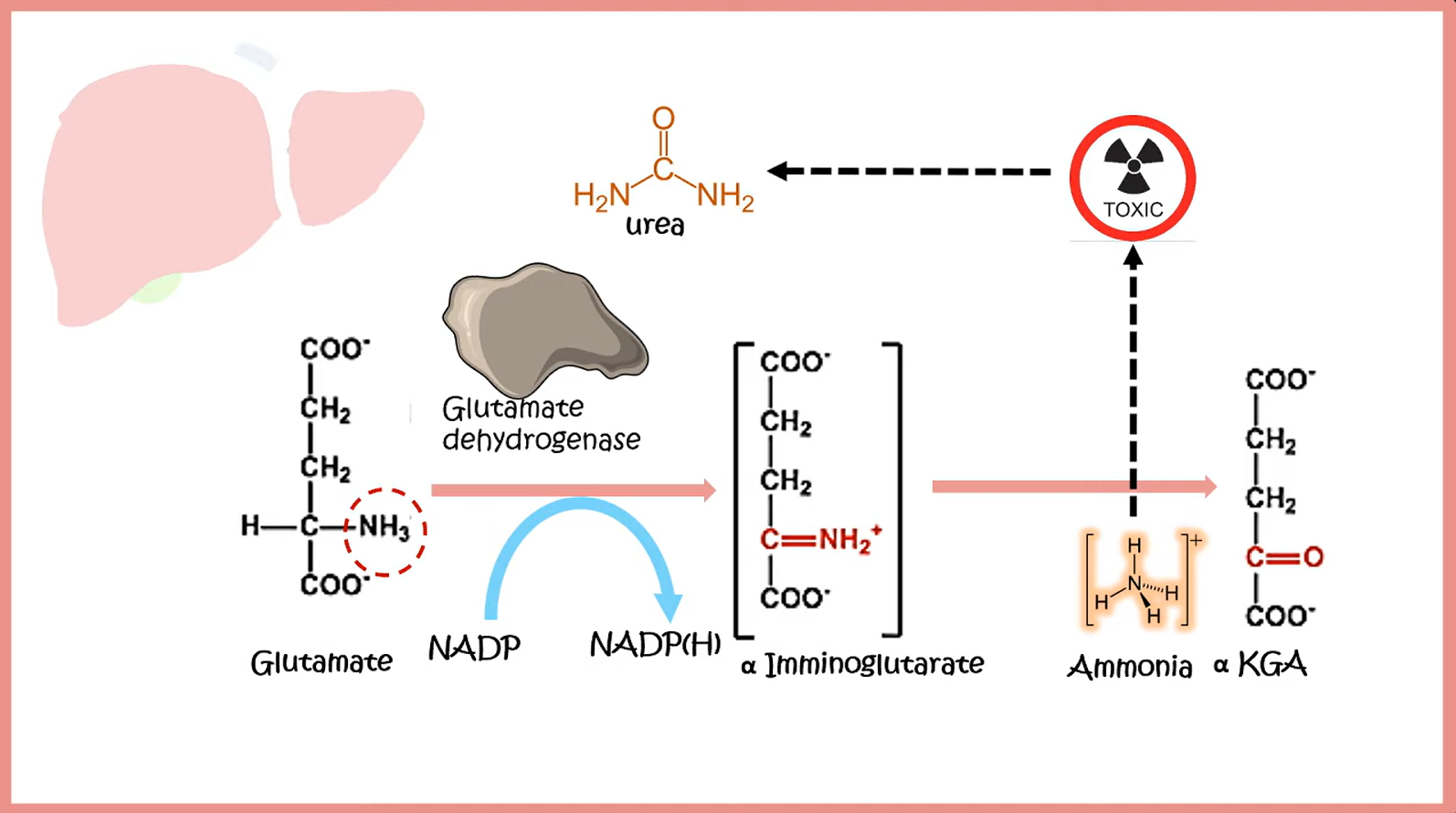
Cahill cycle and Cori cycle

- In the liver, alanine is transaminated by alanine aminotransferase to pyruvate with the amino group being transferred to α-ketoglutarate to form glutamate. Almost all aminotransferase enzymes use α-ketoglutarate as the amino group acceptor.
- Thus, amino groups are funneled into glutamate during protein catabolism.
- Glutamate is further metabolized by the enzyme glutamate dehydrogenase, which liberates free ammonia and regenerates α-ketoglutarate.
- Ammonia then enters the urea cycle to form urea, the primary disposal form of nitrogen in humans.
- Urea subsequently enters the blood and is excreted in the urine.
Cori cycle & Cahill cycle
Lactate/alanine is transported to the liver, where it is converted into glucose. It is then transported back to the muscles for energy production.
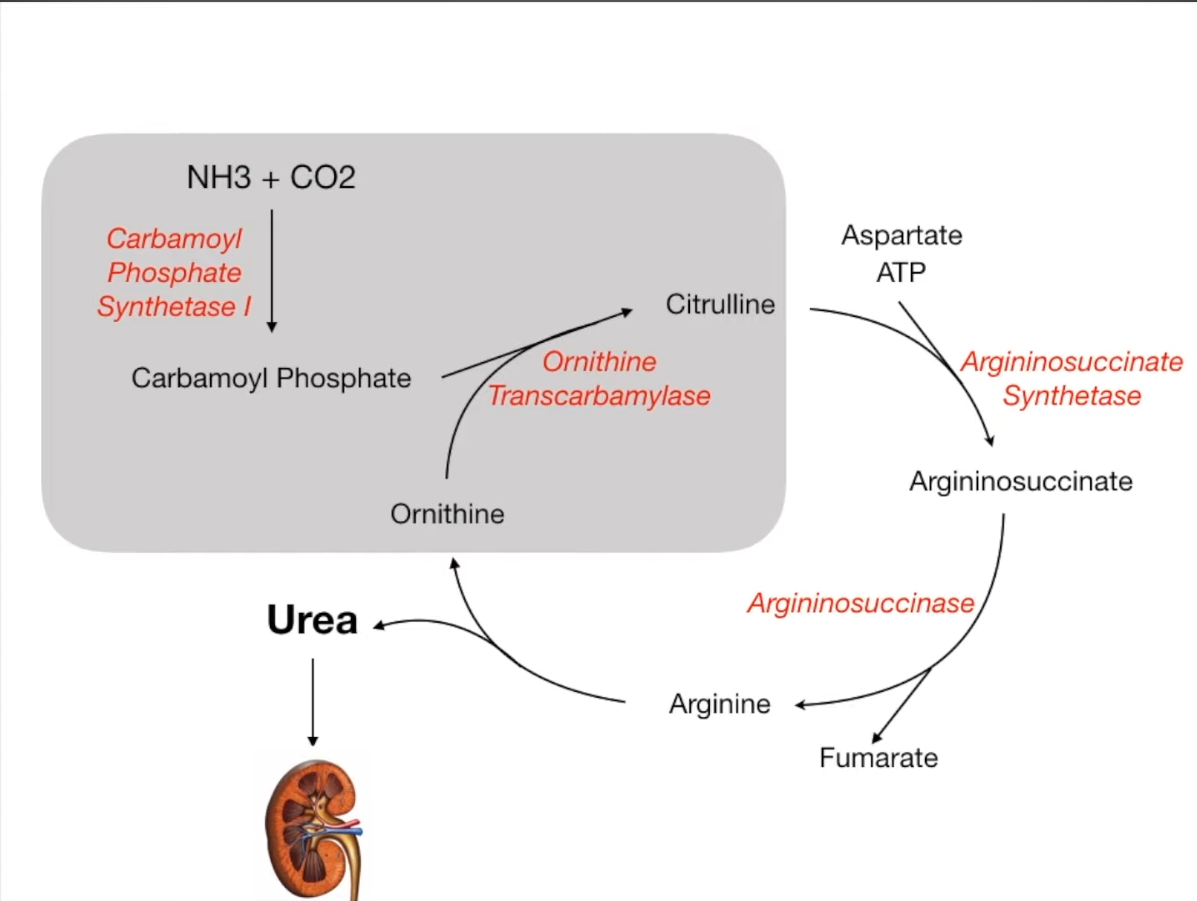
Urea cycle
- Function: Converts ammonia (NH3) into urea for excretion. Occurs in the liver (cytosol and mitochondria).
- Key Steps & Enzymes:
- CO2 + NH3 + 2 ATP → Carbamoyl Phosphate. Enzyme: Carbamoyl Phosphate Synthetase I (CPS I). Rate-limiting step.
- Location: Mitochondria.
- Activator: N-acetylglutamate.
- Carbamoyl Phosphate + Ornithine → Citrulline. Enzyme: Ornithine Transcarbamylase (OTC).
- CO2 + NH3 + 2 ATP → Carbamoyl Phosphate. Enzyme: Carbamoyl Phosphate Synthetase I (CPS I). Rate-limiting step.

Ornithine transcarbamylase deficiency

- Genetics: X-linked Recessive (The only urea cycle disorder not Autosomal Recessive).
- Mechanism: Defect in Ornithine Transcarbamylase → Blocks Carbamoyl phosphate + Ornithine → Citrulline.
- Key Labs:
- ↑ Ammonia
- ↑ Orotic Acid (Excess carbamoyl phosphate shunts to pyrimidine synthesis)
- ↓ Citrulline
- ↓ BUN
- High-Yield Differential:
- vs. Orotic Aciduria: OTC def. has Hyperammonemia and NO megaloblastic anemia.
- vs. CPS1 Deficiency: OTC def. has ↑ Orotic acid (CPS1 def. has low orotic acid).
- Treatment: Low protein diet, nitrogen scavengers.
Arginase deficiency
- Pathophysiology
- Defect: Arginase (Urea Cycle).
- Block: Arginine → Ornithine + Urea.
- Result: Accumulation of Arginine; mild/no hyperammonemia (unlike other UCDs).
- Genetics: Autosomal Recessive.
- Clinical Features
- Spastic diplegia (classic presentation; mimics Cerebral Palsy).
- Choreoathetosis, growth delay, intellectual disability.
- Onset: Toddler/Early childhood (not neonatal).
- Diagnostics
- Labs: Markedly ↑ Plasma Arginine.
- Treatment
- Low protein diet.
- NO arginine supplementation.
Digestion and absorption of dietary proteins
- Mouth: Chewing (mechanical breakdown). No chemical protein digestion.
- Stomach:
- HCl: Denatures proteins and activates pepsinogen to pepsin.
- Pepsin: Breaks proteins into smaller polypeptides.
- Small Intestine (Lumen - major digestion):
- Pancreas releases inactive enzymes (trypsinogen, chymotrypsinogen, etc.).
- Enteropeptidase (from intestinal cells) activates trypsinogen to Trypsin.
- Trypsin then activates other pancreatic enzymes (chymotrypsin, carboxypeptidase).
- These enzymes break polypeptides into smaller peptides (tripeptides, dipeptides) and some free amino acids.
- Small Intestine (Brush Border & Inside Mucosal Cells - final breakdown & absorption):
- Brush border enzymes (aminopeptidases, dipeptidases, tripeptidases) on intestinal cells break small peptides into mostly free amino acids, plus some di- and tripeptides.
- Free amino acids, dipeptides, and tripeptides are absorbed into intestinal mucosal cells (enterocytes).
- Inside enterocytes: Cytosolic peptidases break down remaining di- and tripeptides into free amino acids.
- Bloodstream: Free amino acids are transported from enterocytes into the blood and travel to the liver and then to the rest of the body.