| PSGN | IgA Nephropathy | Alport Syndrome | RPGN (Crescentic) | DPGN (Lupus) | MPGN | |
|---|---|---|---|---|---|---|
| Classic Patient | Child, 2-4 wks post-GAS infx | Young adult, syn-pharyngitic | Boy w/ fam hx of renal failure & deafness | Pt w/ acute, rapid GFR decline | Patient with SLE | Assoc. w/ HCV, cancer, or autoimmune dz |
| Pathophysiology | Type III HSR; IC deposition | Mesangial IgA IC deposition | X-linked defect in Type IV Collagen | GBM breaks → fibrin influx | Type III HSR; diffuse IC deposition | IC deposition or complement dysregulation |
| Key Features / Labs | ↓↓ C3, ↑ASO titer | Normal C3, episodic gross hematuria | Hematuria, sensorineural deafness, ocular defects | Anti-GBM Ab, p/c-ANCA depending on type | ↓↓ C3/C4, +anti-dsDNA | ↓↓ C3 (persistent), +HCV serology |
| LM/IF/EM | Subepithelial humps (EM), hypercellular “lumpy bumpy” (LM), granular IgG/C3 (IF) | Mesangial IgA deposits (IF) | “Basket-weave” GBM (EM) | Crescents (LM); IF defines type (linear, pauci, granular) | “Wire loops” (LM), subendothelial deposits (EM), “full-house” (IF) | “Tram-track” GBM (LM) |
| Key Fact | Prognosis excellent in children | Most common GN worldwide. | ”Can’t see, can’t pee, can’t hear a high C” | A histologic pattern, not a single disease. | Most common & severe nephritis in SLE. | Strong association with Hepatitis C (Type I). |
Etiology
Tip
- The most common cause of nephritic syndrome is immune complex deposition, which leads to serum hypocomplementemia.
- IgA nephropathy is an exception, which is characterized by normal serum complement levels
- ANCA-associated glomerulonephritis (pauci-immune glomerulonephritis)
- Granulomatosis with polyangiitis
- Microscopic polyangiitis
- Eosinophilic granulomatosis with polyangiitis
- Anti-glomerular basement membrane (GBM) associated glomerulonephritis
- Anti-GBM disease (Goodpasture disease)
- Anti-GBM glomerulonephritis (no lung involvement)
- Immune complex-mediated glomerulonephritis
- Low C3 levels
- Lupus nephritis
- Infection-related glomerulonephritis
- Normal C3 levels
- Low C3 levels
Classifications
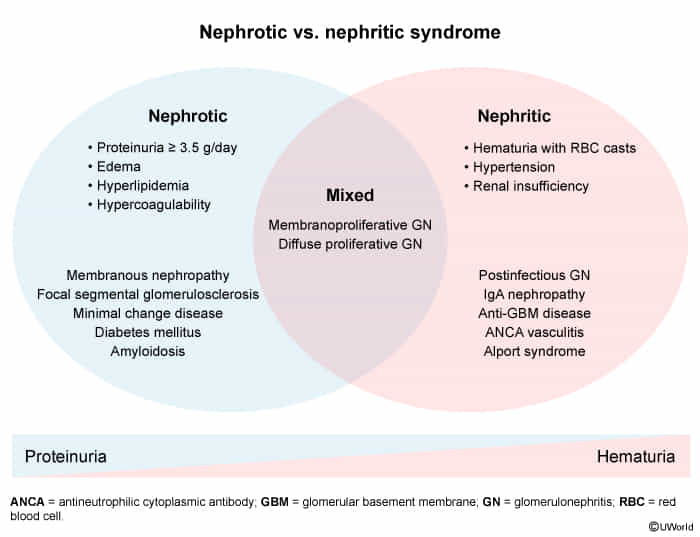
- Poststreptococcal glomerulonephritis
- Diffuse proliferative glomerulonephritis
- Rapidly progressive glomerulonephritis
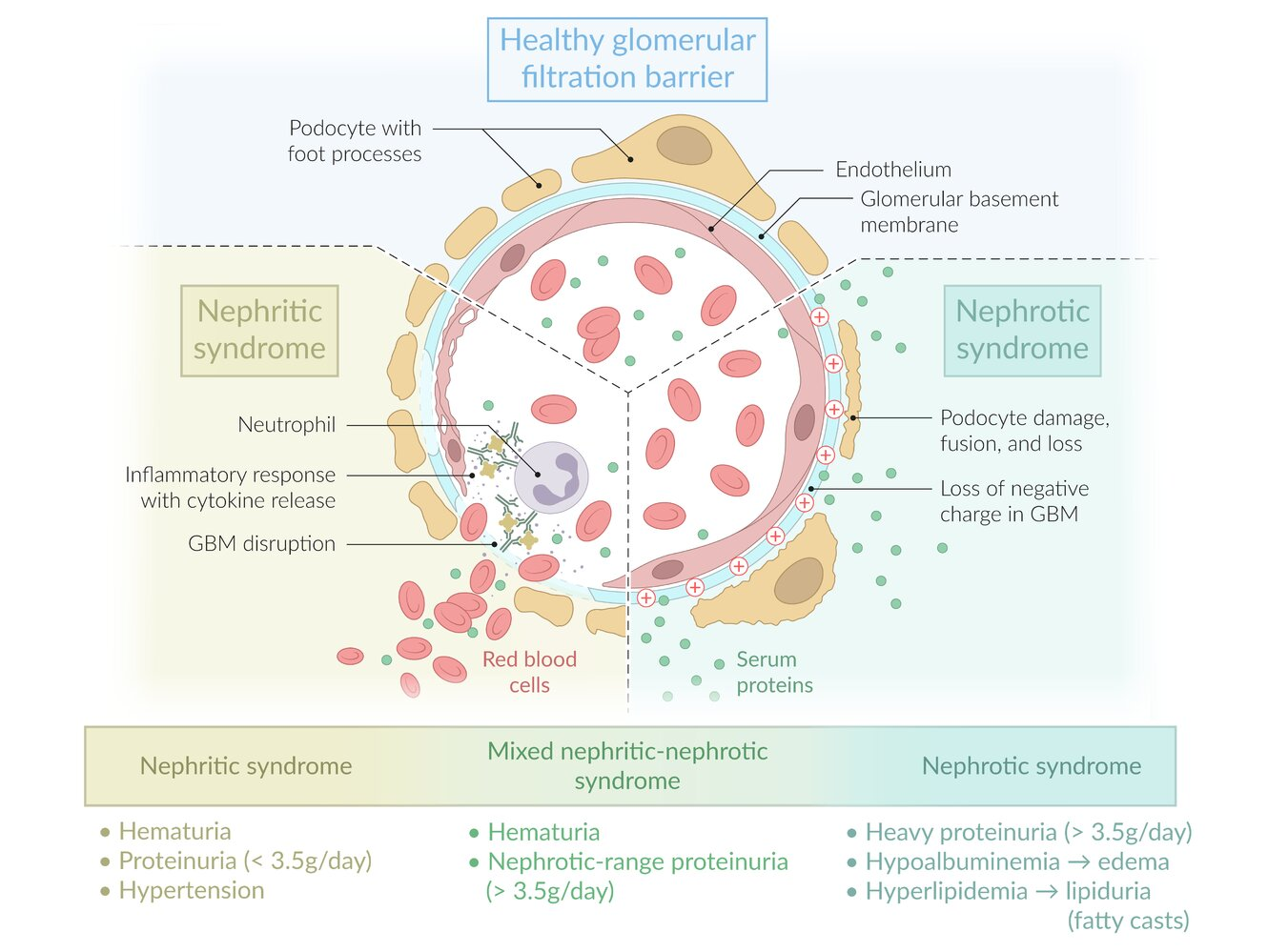
Pathophysiology
Clinical features
Diagnostics

- Initial: Urinalysis (UA) showing microscopic/macroscopic hematuria, dysmorphic RBCs, RBC casts, and sub-nephrotic range proteinuria (< 3.5 g/day).
- Key Labs:
- Renal function: Elevated BUN and serum Cr.
- Serum complements:
- Decreased C3/C4: PSGN, Lupus Nephritis, Membranoproliferative GN (MPGN).
- Normal C3/C4: IgA Nephropathy, Granulomatosis with Polyangiitis (GPA), Microscopic Polyangiitis (MPA), Goodpasture syndrome.
- Serology: Anti-streptolysin O (ASO) or anti-DNase B (PSGN); ANA and anti-dsDNA (SLE); c-ANCA/PR3 (GPA); p-ANCA/MPO (MPA); anti-GBM antibodies (Goodpasture).
- Confirmatory/Gold Standard: Renal biopsy (not always needed in classic pediatric PSGN, but required in adults or progressive cases).
- PSGN: LM shows diffuse hypercellularity; IF shows granular deposition (“lumpy-bumpy” C3/IgG); EM shows subepithelial humps.
- IgA Nephropathy: IF shows IgA-dominant immune complex deposition in the mesangium.
- Goodpasture Syndrome: IF shows linear IgG deposition along the GBM.
- RPGN: LM shows crescents composed of fibrin and macrophages.
- Pauci-immune GN (GPA/MPA): Biopsy shows necrotizing GN with minimal or absent immune complexes on IF.
Treatment
General / Supportive
- Primary Goal: Control volume overload & HTN.
- Methods: Salt/fluid restriction, Loop Diuretics (Furosemide), and ACEi/ARBs (especially with proteinuria).
Disease-Specific Tx
- Post-streptococcal GN (PSGN):
- Supportive care only. Antibiotics treat the strep infection but do not alter the course of the GN.
- IgA Nephropathy (Berger’s):
- ACEi/ARBs are first-line. Add corticosteroids if proteinuria persists (>1g/day) and GFR is preserved.
- Lupus Nephritis (Class III/IV):
- Induction: Corticosteroids + Mycophenolate (MMF) or Cyclophosphamide.
- Maintenance: MMF or Azathioprine.
- ANCA-Associated Vasculitis (e.g., GPA/MPA):
- Induction: Corticosteroids + Rituximab or Cyclophosphamide.
- Anti-GBM Disease (Goodpasture’s):
- Emergency Tx: Plasmapheresis + Corticosteroids + Cyclophosphamide.