- Etiology/Pathophysiology
- Autosomal recessive disorder
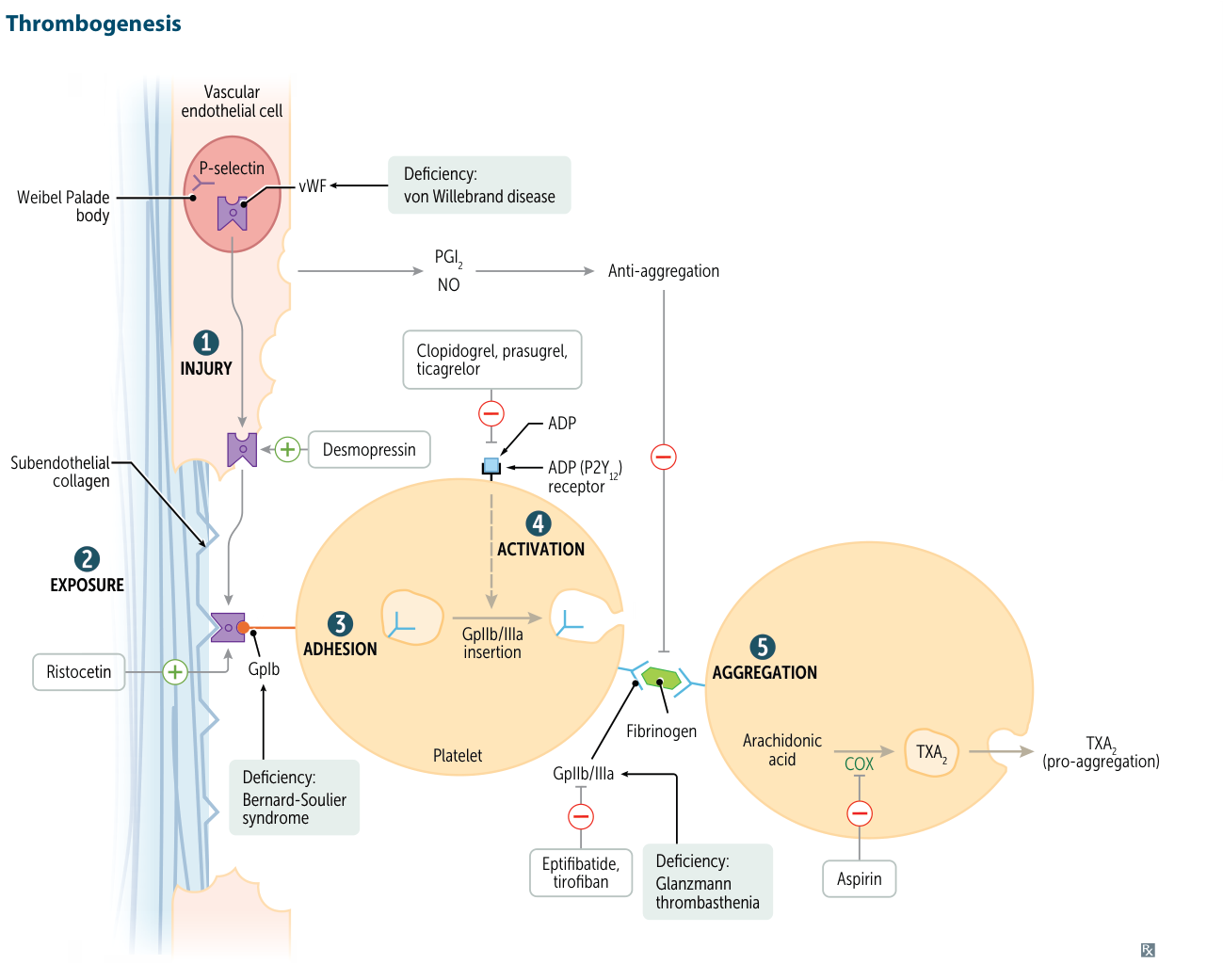
- Deficiency or dysfunction of GPIIb/IIIa (integrin αIIbβ3) on platelet surface
- GPIIb/IIIa normally binds fibrinogen → essential for platelet aggregation
- Platelets cannot aggregate despite normal adhesion (vWF-GPIb interaction intact)
- Clinical Features
- Presents in infancy/early childhood
- Mucocutaneous bleeding: epistaxis, gingival bleeding, menorrhagia, easy bruising
- Severe bleeding with trauma or surgery
- No hepatosplenomegaly or lymphadenopathy
- Diagnostics
- Normal platelet count (key differentiator from ITP)
- Normal PT/PTT (coagulation cascade intact)
- Prolonged bleeding time (or abnormal PFA-100)
- Blood smear: Normal platelet morphology, no platelet clumping
- Abnormal platelet aggregation studies: No aggregation with ADP, collagen, epinephrine, thrombin; normal response to ristocetin (GPIb-vWF interaction intact)
- Flow cytometry: ↓ or absent GPIIb/IIIa expression (definitive Dx)
- Treatment
- Avoid antiplatelet agents (aspirin, NSAIDs)
- Minor bleeding: Local measures, antifibrinolytics (tranexamic acid)
- Major bleeding/surgery: Platelet transfusions (risk of alloimmunization with repeated use)
- Recombinant factor VIIa (rFVIIa) for refractory cases or alloimmunized patients
- Key Associations
- Normal ristocetin response distinguishes from Bernard-Soulier syndrome (abnormal ristocetin, giant platelets, ↓ GPIb)
- Think Glanzmann when: mucocutaneous bleeding + normal platelet count + no aggregation except with ristocetin