Classic Galactosemia (Type I)
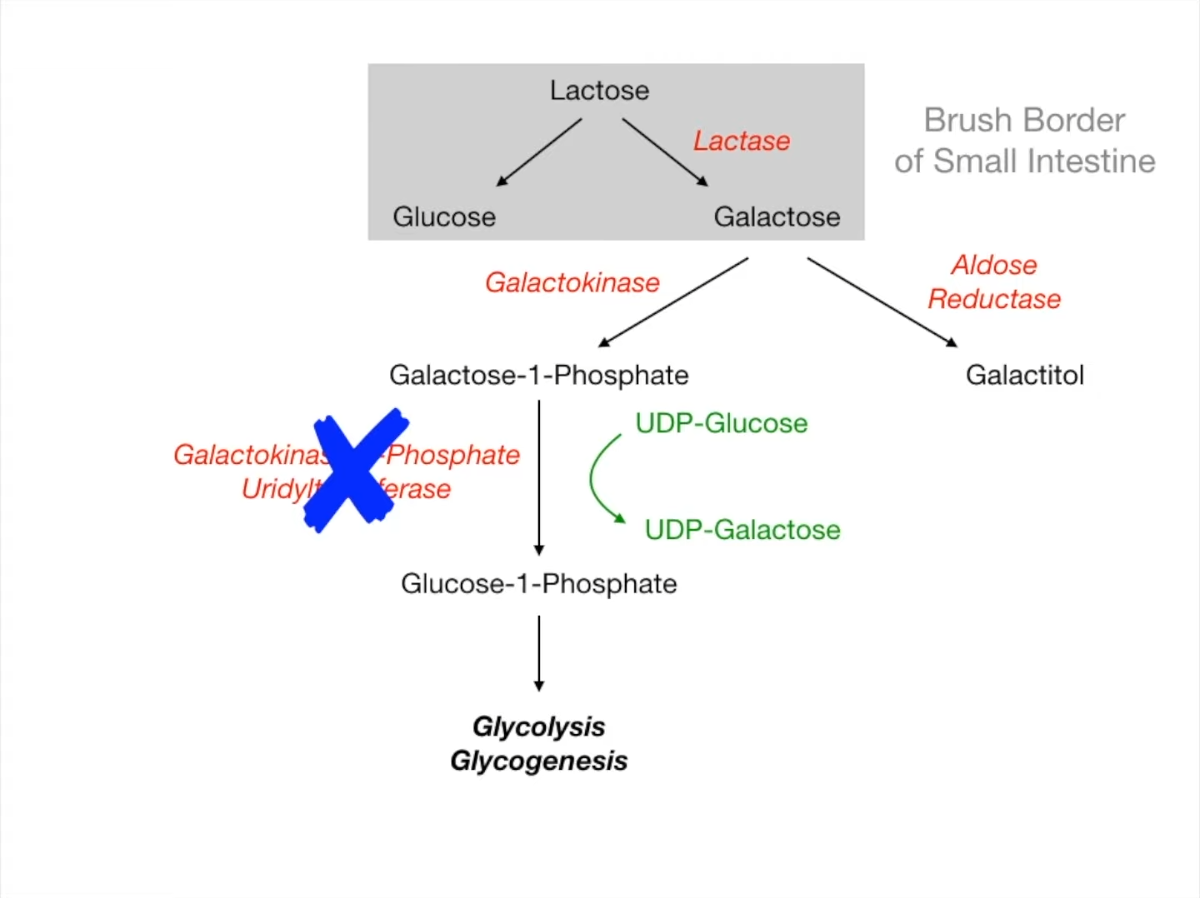
- Enzyme Deficiency: Galactose-1-phosphate uridyltransferase (GALT).
- Inheritance: Autosomal Recessive (AR).
- Pathophysiology:
- Accumulation of Galactose-1-phosphate (toxic to liver/kidney) and Galactitol (lens).
- Phosphate depletion leads to liver failure and renal dysfunction.
- Clinical Features:
- Presents shortly after starting feeding (breast milk or formula).
- Failure to thrive, vomiting, diarrhea.
- Hepatomegaly, jaundice, cirrhosis.
- Infantile Cataracts (due to galactitol accumulation).
- Intellectual disability (if untreated).
- Increased risk of E. coli sepsis.
- Diagnostics:
- Newborn screening (mandatory in US).
- Urine: Non-glucose reducing substances present (Clinitest +).
- Confirmatory: Absence of GALT activity in RBCs.
- Treatment: Strict exclusion of galactose and lactose from diet (soy-based formula).
Tip
- Reducing Substances: Both galactose and glucose are reducing sugars. Urine dipstick tests for glucose specifically (glucose oxidase) and will be negative in galactosemia, but Clinitest detects all reducing sugars and will be positive.
Galactokinase Deficiency (Type II)
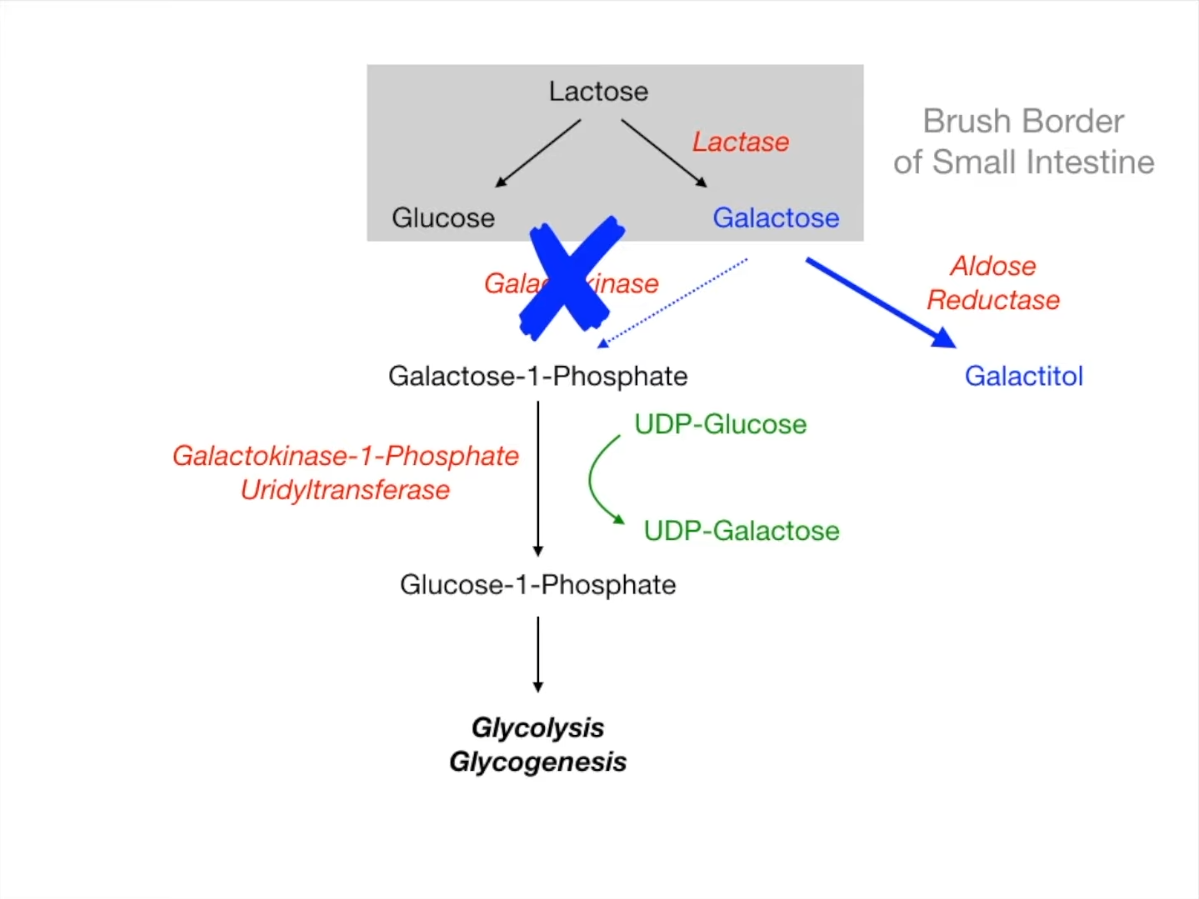
- Enzyme Deficiency: Galactokinase (GALK).
- Inheritance: Autosomal Recessive (AR).
- Pathophysiology:
- Galactose accumulates shunted to Aldose Reductase pathway Galactitol.
- Galactitol is an osmotic agent water influx into lens.
- Clinical Features:
- Milder condition than Classic Galactosemia.
- Infantile Cataracts (often the only manifestation). t
- May present as failure to track objects or develop a social smile.
- No hepatic or renal toxicity (Galactose-1-P does not accumulate).
- Treatment: Dietary restriction of galactose (prevents cataracts).